UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ANNUAL
REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE
SECURITIES EXCHANGE ACT OF 1934
For the
fiscal year ended December 31, 2017
Commission
File Number: 000-08092
GT BIOPHARMA, INC.
(Exact
name of Registrant as specified in its charter)
|
Delaware
|
|
94-1620407
|
|
(State
of incorporation or organization)
|
|
(I.R.S.
Employer Identification No.)
|
1825 K
Street NW, Suite 510
Washington, DC 20006
(Address
of principal executive offices) (Zip code)
(800) 304-9888
(Registrant’s
telephone number including area code)
Securities
registered pursuant to Section 12(b) of the Act: None.
Securities
registered pursuant to section 12(g) of the Act:
|
Title
of Securities
|
|
Exchanges
on which Registered
|
|
Common
Stock, $.001 Par Value
|
|
None
|
Indicate
by check mark if the registrant is a well-known seasoned issuer, as
defined in Rule 405 of the Securities Act. Yes ☐ No
☒
Indicate
by check mark if the registrant is not required to file reports
pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No
☒
Indicate
by check mark whether the registrant (1) has filed all reports
required to be filed by Section 13 or 15(d) of the Securities
Exchange Act of 1934 during the preceding 12 months (or for such
shorter period that the registrant was required to file such
reports), and (2) has been subject to such filing requirements for
the past 90 days. Yes ☒ No ☐
Indicate
by check mark whether the registrant has submitted electronically
and posted on its corporate website, if any, every Interactive Data
File required to be submitted and posted pursuant to Rule 405 of
Regulation S-T during the preceding 12 months (or for such shorter
period that the registrant was required to submit and post such
files). Yes ☐ No ☒
Indicate
by check mark if disclosure of delinquent filers pursuant to Item
405 of Regulation S-K (§229.405) is not contained herein, and
will not be contained, to the best of registrant’s knowledge,
in definitive proxy or information statements incorporated by
reference in Part III of this Form 10-K or any amendment to this
Form 10-K. Yes
☐ No
☒
Indicate
by check mark whether the registrant is a large accelerated filer,
an accelerated filer, a non-accelerated filer, a smaller reporting
company, or emerging growth company. See the definitions of
“large accelerated filer,” “accelerated
filer,” “smaller reporting company,” and
“emerging growth company” in Rule 12b-2 of the Exchange
Act.
|
Large
accelerated filer☐
|
Accelerated
filer ☐
|
|
Non-accelerated
filer ☐ (Do not check if a smaller reporting
company)
|
Smaller
reporting company ☒
|
|
|
Emerging
Growth Company ☐
|
If an
emerging growth company, indicate by check mark if the registrant
has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided
pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant is a shell company (as defined
in Rule 12b-2 of the Exchange Act). Yes ☐ No
☒
The
aggregate market value of the registrant’s common stock,
$0.001 par value per share, held by non-affiliates on June 30, 2017
was approximately $2.5 million. As of February 28, 2018, there were
50,117,978 shares of the registrant’s common stock, $0.001
par value, issued and outstanding.
Table
of Contents
|
PART
I
|
|
|
|
1
|
|
Item
1.
|
|
Business
|
|
1
|
|
Item
1A.
|
|
Risk
Factors
|
|
26
|
|
Item
1B.
|
|
Unresolved
Staff Comments
|
|
48
|
|
Item
2.
|
|
Properties
|
|
48
|
|
Item
3.
|
|
Legal
Proceedings
|
|
48
|
|
Item
4.
|
|
Mine
Safety Disclosures
|
|
48
|
|
|
|
|
|
|
|
PART
II
|
|
|
|
49
|
|
Item
5.
|
|
Market
for Registrant’s Common Equity, Related Stockholder Matters
and Issuer Purchases of Equity Securities
|
|
49
|
|
Item
6.
|
|
Selected
Financial Data
|
|
50
|
|
Item
7.
|
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
|
|
50
|
|
Item
7A.
|
|
Quantitative
and Qualitative Disclosures About Market Risk
|
|
56
|
|
Item
8.
|
|
Financial
Statements and Supplementary Data
|
|
56
|
|
Item
9.
|
|
Changes
in and Disagreements With Accountants on Accounting and Financial
Disclosure
|
|
56
|
|
Item
9A.
|
|
Controls
and Procedures
|
|
56
|
|
Item
9B.
|
|
Other
Information
|
|
57
|
|
|
|
|
|
|
|
PART
III
|
|
|
|
58
|
|
Item
10.
|
|
Directors,
Executive Officers and Corporate Governance
|
|
58
|
|
Item
11.
|
|
Executive
Compensation
|
|
59
|
|
Item
12.
|
|
Security
Ownership of Certain Beneficial Owners and Management and Related
Stockholder Matters
|
|
62
|
|
Item
13.
|
|
Certain
Relationships and Related Transactions, and Director
Independence
|
|
64
|
|
Item
14.
|
|
Principal
Accounting Fees and Services
|
|
64
|
|
|
|
|
|
|
|
PART
IV
|
|
|
|
65
|
|
Item
15.
|
|
Exhibits,
Financial Statement Schedules
|
|
65
|
PART I
CAUTIONARY NOTICE REGARDING FORWARD-LOOKING STATEMENTS
This
Report, including any documents which may be incorporated by
reference into this Report, contains “Forward-Looking
Statements” within the meaning of Section 27A of the
Securities Act of 1933, as amended, and Section 21E of the
Securities Exchange Act of 1934, as amended. All statements other
than statements of historical fact are “Forward-Looking
Statements” for purposes of these provisions, including our
plans of operation, any projections of revenues or other financial
items, any statements of the plans and objectives of management for
future operations, any statements concerning proposed new products
or services, any statements regarding future economic conditions or
performance, and any statements of assumptions underlying any of
the foregoing. All Forward-Looking Statements included in this
document are made as of the date hereof and are based on
information available to us as of such date. We assume no
obligation to update any Forward-Looking Statement. In some cases,
Forward-Looking Statements can be identified by the use of
terminology such as “may,” “will,”
“expects,” “plans,”
“anticipates,” “intends,”
“believes,” “estimates,”
“potential,” or “continue,” or the negative
thereof or other comparable terminology. Although we believe that
the expectations reflected in the Forward-Looking Statements
contained herein are reasonable, there can be no assurance that
such expectations or any of the Forward-Looking Statements will
prove to be correct, and actual results could differ materially
from those projected or assumed in the Forward-Looking Statements.
Future financial condition and results of operations, as well as
any Forward-Looking Statements are subject to inherent risks and
uncertainties, including any other factors referred to in our press
releases and reports filed with the Securities and Exchange
Commission. All subsequent Forward-Looking Statements attributable
to the company or persons acting on its behalf are expressly
qualified in their entirety by these cautionary statements.
Additional factors that may have a direct bearing on our operating
results are described under “Risk Factors” and
elsewhere in this report.
Introductory Comment
Throughout
this Annual Report on Form 10-K, the terms “GTBP,”
“we,” “us,” “our,” “the
company” and “our company” refer to GT Biopharma,
Inc., a Delaware corporation formerly known as DDI Pharmaceuticals,
Inc., Diagnostic Data, Inc. and Oxis International, Inc., together
with our subsidiaries.
ITEM
1. BUSINESS
We are a clinical stage
biopharmaceutical company focused on the development and
commercialization of novel immuno-oncology products based off our
proprietary Tri-specific Killer Engager (TriKE), Tetra-specific
Killer Engager (TetraKE) and bi-specific Antibody Drug Conjugate
(ADC) technology platforms. Our TriKE and TetraKE platforms
generate proprietary moieties designed to harness and enhance the
cancer killing abilities of a patient’s own natural killer,
or NK, cells. Once bound to a NK cell, our moieties are designed to
enhance the NK cell and precisely direct it to one or more
specifically-targeted proteins (tumor antigens) expressed on a
specific type of cancer, ultimately resulting in the cancer
cell’s death. TriKEs and TetraKEs are made up of recombinant
fusion proteins, can be designed to target any number of tumor
antigens on hematologic malignancies, sarcomas or solid tumors and
do not require patient-specific customization. They are designed to
be dosed in a common outpatient setting similar to modern antibody
therapeutics and are expected to have reasonably low cost of goods.
Our ADC platform generates product candidates that are bi-specific,
ligand-directed single-chain fusion proteins that, we believe,
represent the next generation of ADCs.
Our most advanced bi-specific ADC, which targets CD19+ and/or CD22+
hematological malignancies, is in a Phase 2 NHL/ALL trial, and we
plan to begin a Phase 1 trial in CD33+ hematologic malignancies for
our most advanced TriKE product candidate in the second half of
2018. We are initially targeting certain hematologic malignancies
as we believe our product candidates may have certain advantages
over existing and other in-development products. We are also
developing TetraKE product candidates designed to target the larger
solid tumor market and expect to begin human clinical trials in
2019.
We also are focused on developing a portfolio of three central
nervous system, or CNS, product candidates that are covered by
issued or filed composition of matter patents and consist of
innovative reformulations and/or repurposing of existing therapies.
We expect to take advantage of our CNS portfolio by generating
proof-of-concept data and/or achieving other milestones and
ultimately entering into strategic transactions, which may include
transactions with commercialization-oriented pharmaceutical
companies.
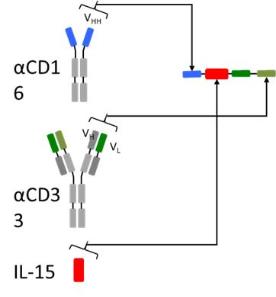
Our TriKE product candidates are single-chain, tri-specific scFv
recombinant fusion proteins composed of the variable regions of the
heavy and light chains (or heavy chain only) of anti-CD16
antibodies, wild-type or a modified form of IL-15 and the variable
regions of the heavy and light chains of an antibody that precisely
targets a specific tumor antigen. We utilize the NK stimulating cytokine
human IL-15 as a crosslinker between the two scFvs which provides a
self-sustaining signal that leads to the proliferation and
activation of NK cells thus enhancing their ability to kill cancer
cells mediated by antibody-dependent
cell-mediated cytotoxicity (ADCC) via the highly potent CD16
activating receptor on our moieties. Our lead TriKE, OXS-3550,
targeting CD33+ malignancies is expected to begin clinical testing
in the second half of 2018. Our second TriKE product candidate,
OXS-C3550, is a next-generation version of OXS-3550 containing a
modified CD16 component.
Our TetraKE product candidates are single-chain fusion proteins
composed of human single-domain anti-CD16 antibody, wild-type IL-15
and the variable regions of the heavy and light chains of two
antibodies that target two specific tumor antigens expressed
on specific types of cancer cells. An
example of a TetraKE product candidate is OXS-1615which targets
EpCAM and CD133 positive solid tumors. EpCAM is found on many solid
tumor cells of epithelial origin and CD133 is a marker for cancer
stem cells. OXS-1615 is designed to enable a patient’s NK
cells to kill not only the heterogeneous population of cancer cells
found in many solid tumors but also kill the cancer stem cells that
are typically responsible for recurrences. We intend to initiate
human clinical testing for certain of our solid tumor product
candidates in 2019.
Our TriKEs and TetraKEs act by binding to a patient’s NK cell
and a specific tumor antigen enabling an immune synapse between the
now IL-15-enhanced NK cell and the targeted cancer cell. The
formation of this immune synapse induces NK cell activation leading
to the death of the cancer cell. The self-sustaining signal caused
by our IL-15 cross-linker enables prolonged and enhanced
proliferation and activation of NK cells similar to the increased
proliferation of T-cells caused by 41BB-L or CD28 intracellular
domains in CAR-T therapy but without the need to enhance the
patient’s NK cells ex vivo.
We are using our TriKE and TetraKE platforms with the intent to
bring to market multiple immuno-oncology products that can treat a
wide range of hematologic malignancies, sarcoma and solid tumors.
The platforms are scalable and we are putting processes in place to
be able to produce IND-ready moieties in approximately 90-120 days
after a specific TriKE or TetraKE conceptual design. After
conducting market and competitive research, specific moieties can
then be rapidly advanced into the clinic on our own or through
potential collaborations with larger companies. We are currently
evaluating over a dozen moieties and intend to announce additional
clinical product candidates in the second half of 2018. We believe
our TriKEs and TetraKEs will have the ability, if approved for
marketing, to be used on a stand-alone basis, augment the current
monoclonal antibody therapeutics, be used in conjunction with more
traditional cancer therapy and potentially overcome certain
limitations of current chimeric antigen receptor, or CAR-T,
therapy.
We also believe our bi-specific, ligand-directed single-chain
fusion proteins represents the next generation of ADCs. Our lead
bi-specific ADC, OXS-1550, which targets CD19+ and/or CD22+
hematological malignancies is currently in a Phase 2 trial being
conducted at the University of Minnesota Masonic Cancer Center in
patients with relapsed/refractory B-cell leukemias or lymphomas. We
believe OXS-1550 has certain properties that could result in
competitive advantages over recently approved ADC products
targeting leukemias and lymphomas. In a Phase 1 trial, of nine
patients that achieved adequate blood levels, we saw a durable
complete response, or CR, in two heavily pretreated patients. One
patient, who had failed multiple previous treatment regimens, has
been cancer free since early 2015.
Our initial work has been conducted in collaboration with the
Masonic Cancer Center at the University of Minnesota under a
program led by Dr. Jeffrey Miller, the Deputy Director. Dr. Miller
is a recognized leader in the field of NK cell and IL-15 biology
and their therapeutic potential. We have exclusive rights to the
TriKE and TetraKE platforms and are generating additional
intellectual property around specific moieties.
We also are focused on developing a portfolio of central nervous
system, or CNS, product candidates that are covered by formulation
patents and issued or filed composition of matter patents and
consist of innovative reformulations and/or repurposing of existing
therapies. We believe our CNS product candidates represent
potentially near-to-market opportunities that may have broader
potential applicability beyond their initial indication. Certain
members of our management team have a track-record of developing
CNS products and product candidates with similar strategies
including at Chase Pharmaceuticals and Prestwick Pharmaceuticals.
We have designed our CNS clinical programs with the intent to
efficiently advance each program to an FDA New Drug Application in
a certain initial indication and expand applicable markets
potentially after approval. Our three product candidates are
initially targeting a rare autoimmune disease Myasthenia Gravis, a
rare neuropathic pain indication, trigeminal neuralgia, and a
vestibular disorder, motion sickness.
Immuno-Oncology Platform
Tri-specific Killer Engagers (TriKEs) and Tetra-specific Killer
Engagers (TetraKEs)
The generation of chimeric antigen receptor, or CAR, expressing T
cells from monoclonal antibodies has represented an important step
forward in cancer therapy. These therapies involve the genetic
engineering of T cells to express either CARs, or T cell receptors,
or TCRs, and are designed such that the modified T cells can
recognize and destroy cancer cells. While a great deal of interest
has recently been placed upon chimeric antigen receptor T, or
CAR-T, therapy, it has certain limitations for broad potential
applicability because it can require an individual approach that is
expensive and time consuming, and may be difficult to apply on a
large scale. We believe there is an unmet need for targeted
immuno-oncology therapies that have the potential to be dosed in a
patient-friendly outpatient setting, can be used on a stand-alone
basis, augment the current monoclonal antibody therapeutics and/or
be used in conjunction with more traditional cancer therapy. We
believe our TriKE and TetraKE constructs have this potential and
therefore we have generated, and intend to continue to generate, a
pipeline of product candidates to be advanced into the clinic on
our own or through potential collaborations with larger
companies.
NK cells represent an important immunotherapeutic target as they
are involved in tumor immune-surveillance, can mediate
antibody-dependent cell-mediated cytotoxicity (ADCC), contain
pre-made granules with perforin and granzyme B and can quickly
secrete inflammatory cytokines, and unlike T cells they do not
require antigen priming and can kill cells in the absence of major
histocompatibility complex (MHC) presentation.
Unlike full-length antibodies, TriKEs and TetraKEs are small
single-chain fusion proteins that bind the CD16 receptor of NK
cells directly producing a potent and lasting response, as
demonstrated by preclinical studies. An additional benefit they may
have is attractive biodistribution, as a consequence of their
smaller size, which we expect to be important in the treatment of
solid tumors. In addition to these advantages, TriKEs and TetraKEs
are designed to be non-immunogenic, have appropriate clearance
properties and can be engineered quickly to target a variety of
tumor antigens.
Background and Select Non-Clinical Data
In conjunction with our research agreement with the Masonic Cancer
Center at the University of Minnesota, the exploration of targeting
NK cells to a variety of tumors initially focused on novel
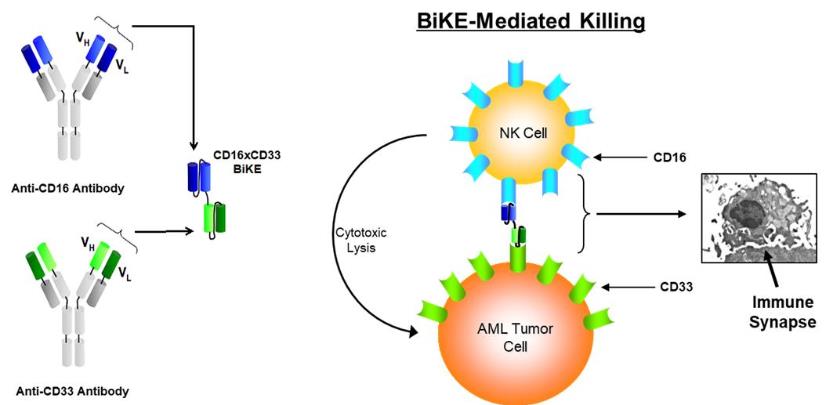
bi-specific killer engagers, or BiKEs, composed of the variable
portions of antibodies targeting the CD16 activating receptor on NK
cells and CD33 (AML and MDS; see figure below), CD19/CD22 (B cell
lymphomas), or EpCAM (epithelial tumors (breast, colon, and lung))
on the tumor cells.
Subsequently, a tri-specific (TriKE) construct that replaced the
linker molecule between the CD16 scFv and the CD33 scFv with a
modified IL-15 molecule, containing flanking sequences, was
generated and tested. Data indicate that the CD16 x IL-15 x CD33
and CD16 x IL-15 x EpCAM TriKEs potently induce proliferation of
healthy donor NK cells, possibly greater than that induced by
exogenous IL-15, which is absent in the BiKE platform. Targeted
delivery of the IL-15 through the TriKE also resulted in specific
expansion of the NK cells without inducing T cell expansion on
post-transplant patient samples.
When compared to the CD16 x CD33 BiKE, the CD16 x IL-15 x CD33
TriKE is also capable of potently restoring killing capacity of
post-transplant NK cells against CD33-expressing HL-60 Targets and
primary AML blasts. These results demonstrated the ability to
functionally incorporate an IL-5 cytokine into the BiKE platform
and also demonstrated the possibility of targeting a variety of
cytokines directly to NK cells while reducing off-target effects
and the amount of cytokines needed to obtain biologically relevant
function.
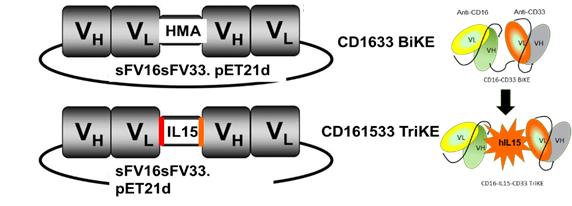
The figure below is a schematic of a BiKE construct (top) and a
TriKE construct (bottom), which has the modified IL-15 linker
between the CD16 scFv and the CD33 scFv components.
The TriKE constructs were also tested against three separate human
tumor cell lines: HL-60 (promyelocitic leukemia), Raji
(Burkitt’s lymphoma), and HT29 (colorectal adenocarcinoma),
in addition to a model for ovarian cancer. All cell lines contained
the Luc reporter to allow for in vivo imaging of the tumors. These
systems were used to show in vivo efficacy of BiKEs (1633) and
TriKEs (OXS-3550) against relevant human tumor targets (HL-60-luc)
over an extended period of time. The system consisted of initial
conditioning of mice using radiation (250-275 cGy), followed by
injection of the tumor cells (I.V. for HL-60-luc and Raji-luc,
intra-splenic for HT29-luc and IP for ovarian for MA-148-luc), a
three-day growth phase, injection of human NK cells, and repeated
injection of the drugs of interest, BiKE and TriKE (three to five
times a week). Imaging was carried out at day 7, 14, and 21, and
extended as needed.
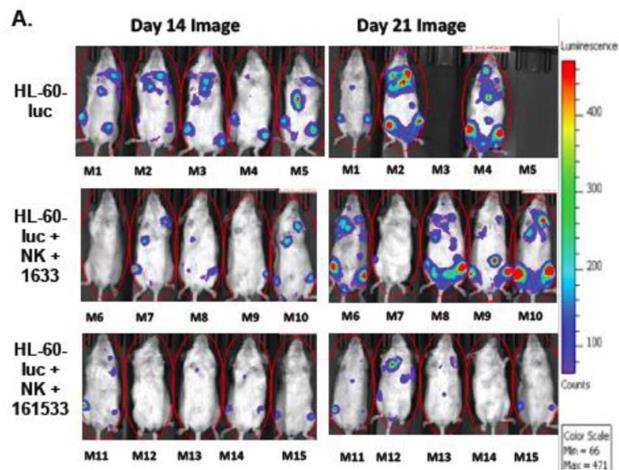
Figure A below shows the results (tumor burden and mortality) when
dosing NK cells alone (top panel), the BiKE version (lacking IL-15)
of OXS-3550 (middle panel; called 1633), and the TriKE, OXS-3550
(bottom panel; then called 161533) in the above described human
tumor model, HL-60-luc. In the NK-cell-only arm, two out of the
five mice were dead by day 21 with two of the surviving mice having
extensive tumor burden as depicted by the colored images. In
contrast, all five mice in each of the BiKE and TriKE arms
survived. In addition, the tumor burden in the TriKE-treated mice
was significantly less than in the BiKE-treated mice, demonstrating
the improved efficacy from NK cells in the TriKE-treated
mice.
Based on these results, and others, the IND for OXS-3550 was filed
in June 2017 by the University of Minnesota. FDA requested that
additional nonclinical toxicology be conducted prior to initiating
clinical trials. The FDA also requested some additional information
and clarifications on the manufacturing (CMC) and clinical
packages. We plan to incorporate the requested additional
toxicology studies, information and clarifications in
the IND that was transferred to us from the University of
Minnesota in October 2017. We expect to begin a Phase 1 clinical
trial for OXS-3550 in the second half of 2018.
Generation of humanized single-domain antibody targeting CD16 for
incorporation into the TriKE platform
To develop second generation TriKEs, we designed a new humanized
CD16 engager derived from a single-domain antibody. While scFvs
consist of a heavy and a light variable chain joined by a linker,
single-domain antibodies consist of a single variable heavy chain
capable of engaging without the need of a light chain counterpart
(see figure below).
These single-domain antibodies are thought to have certain
attractive features for antibody engineering, including physical
stability, ability to bind deep grooves, and increased production
yields, amongst others. Pre-clinical studies demonstrated increased
activity (NK Cell Degranulation) and functionality (NC Cell
Cytokine Production) of the single-domain CD16 TriKE (OXS-C3550)
compared to the original TriKE (OXS-3550) (see figure below). These
data were presented at the 2017 American Society of Hematology
Conference.
Targeting Solid Tumors and Other Potentially Attractive
Characteristics
Unlike full-length antibodies, TriKEs and TetraKEs are small
single-chain fusion proteins that bind the CD16 receptor of NK
cells directly producing a potentially more potent and lasting
response as demonstrated by preclinical studies. An additional
benefit that they may have is an attractive biodistribution,
because of their smaller size, which we expect to be important in
the treatment of solid tumors. In addition to these potential
advantages, TriKEs and TetraKEs are designed to be non-immunogenic,
have appropriate clearance properties and can be engineered quickly
to target a variety of tumor antigens. We believe these attributes
make them an ideal pharmaceutical platform for potentiated NK
cell-based immunotherapies and have the potential to overcome some
of the limitations of CAR-T therapy and other antibody
therapies.
Examples of our earlier stage solid tumor targeting product
candidates are focused on EpCAM, Her2, Mesothelin (mesothelioma and
lung adenocarcinoma), and CD133 alone and in combination. We
believe certain of these constructs have the potential to target
prostate, breast, colon, ovarian, liver, and head and neck cancers.
We intend to initiate human clinical testing for certain of our
solid tumor product candidates in 2019.
Efficient Advancement of Potential Future Product Candidates
--Production and Scale Up
We are using our TriKE and TetraKE platforms with the intent to
bring to market multiple immuno-oncology products that can treat a
range of hematologic malignancies, sarcomas and solid tumors. The
platforms are scalable and we are currently working with several
third parties investigating the optimal expression system of the
TriKEs and TetraKE constructs which we expect to be part of a
process in which we are able to produce IND-ready moieties in
approximately 90-120 days after the construct conceptual
design.
After conducting market and competitive research, specific moieties
can then be rapidly advanced into the clinic on our own or through
potential collaborations with larger companies. We are currently
evaluating over a dozen moieties and intend to announce additional
clinical product candidates in the second half of
2018.
We believe our TriKEs and TetraKEs will have the ability, if
approved for marketing, to be used on a stand-alone basis, augment
the current monoclonal antibody therapeutics, or be used in
conjunction with more traditional cancer therapy and potentially
overcome certain limitations of current chimeric antigen receptor,
or CAR-T, therapy.
Bi-specific Antibody-Drug Conjugates Program
Antibody–drug conjugates (ADCs) are a class of potent
biopharmaceutical drugs designed as a targeted therapy for the
treatment of cancer. ADCs combine the antitumor potency
of highly cytotoxic small-molecule drugs with the high selectivity,
pharmacokinetic profile of mAbs. These attributes allow sensitive
discrimination between healthy and diseased tissue. We believe our
bi-specific, ligand-directed single-chain fusion protein represents
an example of the next generation of ADCs.
OXS-1550, our bi-specific ADC, is a single chain bispecific
recombinant fusion protein consisting of an anti-CD22 sFv, an
anti-CD19 sFv, and DT390 (the catalytic and translocation domains
of diphtheria toxin). It is a cytotoxic molecule produced by
recombinant DNA techniques composed of a fusion gene consisting of
sequences for DT390 and also sequences encoding two separate and
distinct sFvs, one recognizing CD22 and one recognizing CD19. The
anti-CD22 sFv comes from the monoclonal antibody RFB4 and this sFv
is currently in clinical trials involving another anti-CD22
immunotoxin called BL22. The anti-CD19 sFv is from the monoclonal
antibody HD37 that has previously been used clinically. Published
preclinical studies have shown that the presence of both sFvs on
the same single chain molecule results in a bispecific fusion toxin
that has superior activity and anti-cancer effects compared to the
monospecific fusion toxins. Between the VL and VH regions of the
sFvs, we have introduced aggregation reducing sequences (ARL) which
has produced a product which has demonstrated better activity
against scid mouse systemic models of B cell malignancy. The action
of DT2219 occurs as a result of binding to the CD22 and/or CD19
receptors, subsequent internalization, and enzymatic inhibition of
protein synthesis leading to cell death.
We believe that our single-chain bi-specific recombinant fusion
proteins utilizing novel linkers and innovative warheads represent
an important advance over currently marketed ADCs. Utilizing our
bi-specific ADC platform we have the ability to generate novel ADCs
with unique targets, linkers and warheads. This platform provides
us with the ability to rapidly construct novel ADCs with the
potential to treat a wide range of cancers, including hematologic
and solid tumors.
Immuno-Oncology Product Candidates
OXS-1550
OXS-1550 is a bispecific scFv recombinant fusion protein-drug
conjugate composed of the variable regions of the heavy and light
chains of anti-CD19 and anti-CD22 antibodies and a modified form of
diphtheria toxin (DT390) as its cytotoxic drug payload. CD19
is a membrane glycoprotein present on the surface of all stages of
B-lymphocyte development and is also expressed on most B-cell
mature lymphoma cells and leukemia cells. CD22 is a
glycoprotein expressed on B-lineage lymphoid precursors, including
precursor acute lymphoblastic leukemia, and often is co-expressed
with CD19 on mature B-cell malignancies such as
lymphoma.
OXS-1550 targets cancer cells expressing the CD19 receptor or CD22
receptor or both receptors. When OXS-1550 binds to cancer
cells, the cancer cells internalize OXS-1550, and are killed due to
the action of drug’s cytotoxic diphtheria toxin
payload. OXS-1550 has completed a Phase 1 human clinical
trial in patients with relapsed/refractory B-cell lymphoma or
leukemia.
The initial Phase 1 study enrolled 25 patients with mature or
precursor B-cell lymphoid malignancies expressing
the CD19 receptor or CD22 receptor or
both receptors. All 25 patients
received at least a single course of therapy. The treatment at the
higher doses produced objective tumor responses with one patient in
continuous partial remission and the second in complete remission.
A Phase 2 trial of OXS-1550 is underway in patients with ALL/NHL.
The FDA-approved clinical trial is being conducted at the
University of Minnesota's Masonic Cancer Center. There are
currently 18 patients enrolled in this clinical trial. Patients in
this trial are given an approved increased dosage and schedule of
OXS-1550.
We began enrolling patients in Phase 2 trial of OXS-1550 during the
first quarter of 2017 and the first patient began dosing in April
2017. We expect data from this
Phase 2 trial to be available in the second half
2018.
OXS-3550
OXS-3550 is our first TriKE product candidate. It is a
single-chain, tri-specific scFv recombinant fusion protein
conjugate composed of the variable regions of the heavy and light
chains of anti-CD16 and anti-CD33 antibodies and a modified form of
IL-15. We intend to study this anti-CD16-IL-15-anti-CD33
TriKE in CD33 positive leukemias, a marker expressed on tumor cells
in acute myelogenous leukemia, or AML, myelodysplastic syndrome, or
MDS, and other hematopoietic malignancies. CD33 is primarily a
myeloid differentiation antigen with endocytic properties broadly
expressed on AML blasts and, possibly, some leukemic stem cells.
CD33 or Siglec-3 (sialic acid binding Ig-like lectin 3, SIGLEC3,
SIGLEC3, gp67, p67) is a transmembrane receptor expressed on cells
of myeloid lineage. It is usually considered myeloid-specific, but
it can also be found on some lymphoid cells. The anti-CD33 antibody
fragment that will be used for these studies was derived from the
M195 humanized anti-CD33 scFV and has been used in multiple human
clinical studies. It has been exploited as target for therapeutic
antibodies for many years. We believe the recent approval of the
antibody-drug conjugate gemtuzumab validates this targeted
approach.
The
OXS-3550 IND will focus on AML, the most common form of adult
leukemia with 21,000 new cases expected in 2018 alone (American
Cancer Society). These patients typically receive frontline
therapy, usually chemotherapy, including cytarabine and an
anthracycline, a therapy that has not changed in over 40 years.
About half will have relapses and require alternative therapies. In
addition, MDS incidence rates have dramatically increased in the
population of the United States from 3.3 per 100,000 individuals
from 2001-2004 to 70 per 100,000 annually, MDS is especially
prevalent in elderly patients that have a median age of 76 years at
diagnosis. The survival of patients with MDS is poor due to
decreased eligibility, as a result of advanced age, for allogeneic
hematopoietic cell transplantation (Allo-HSCT), the only curative
MDS treatment (Cogle CR. Incidence and Burden of the
Myelodysplastic Syndromes. Curr
Hematol Malig Rep. 2015; 10(3):272-281). We expect OXS-3550
could serve as a relatively safe, cost-effective, and easy-to-use
therapy for resistant/relapsing AML and could also be combined with
chemotherapy as frontline therapy thus targeting the larger
market.
The IND for OXS-3550 was filed in June 2017 by the University of
Minnesota. FDA requested that additional preclinical toxicology be
conducted prior to initiating clinical trials. The FDA also
requested some additional information and clarifications on the
manufacturing (CMC) and clinical packages. The
requested additional toxicology studies, information
and clarifications will be incorporated by our company in
the IND that was transferred to us from the University of
Minnesota in October 2017. We expect to begin a Phase 1 clinical
trial in the second half of 2018.
OXS-C3550
OXS-C3550 is a next-generation, follow-on, to our lead TriKE,
OXS-3550. OXS-C3550 contains a modified CD16 moiety which has
improved binding characteristics and enhanced tumor cell killing
based on functional assays and animal models of AML. Using our
platform technology, we substituted the anti-CD16 scFv arm in
OXS-3550 with a novel humanized single-domain anti-CD16 antibody to
create this second-generation molecule which may have improved
functionality. Single-domain antibodies, such as OXS-C3550,
typically have several advantages, including better stability and
solubility, more resistance to pH changes, can better recognize
hidden antigenic sites, lack of a VL
portion thus preventing
VH/VL
mispairing and are suitable for
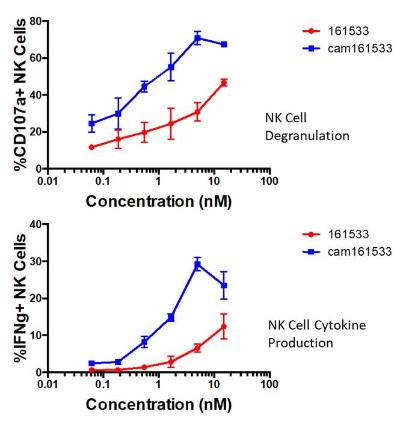
construction of larger molecules. OXS-C3550 induced a potent
increase in NK cell degranulation, measured by CD107a expression
against HL-60 AML tumor targets when compared to our
first-generation TriKE (70.75±3.65% vs. 30.75±5.05%).
IFN production was similarly enhanced (29.2±1.8%
vs. 6.55±1.07%). OXS-C3550 also exhibited a robust increase in
NK cell proliferation (57.65±6.05% vs.
20.75±2.55%).
OXS-3550 studies will help inform the development of OXS-C3550
which we expect will de-risk the OXS-C3550 program as data will be
generated to make an informed decision on which, or both, will be
brought into later phase studies.
OXS-1615
OXS-1615 is an example of our first-generation TetraKEs designed
for the treatment of solid tumors. It is a single-chain fusion
protein composed of CD16-IL15-EpCAM-CD133. EpCAM is found on many
solid tumor cells of epithelial origin and CD133 is a marker for
cancer stem cells. This TetraKE is designed to target not only the
heterogeneous population of cancer cells found in solid tumors but
also the cancer stem cells that are typically responsible for
recurrences. We intend to initiate human clinical testing for
certain of our solid tumor product candidates in 2019.
Central Nervous System
Our CNS portfolio consists of innovative reformulations and/or
repurposing of existing therapies and are covered by issued
formulation or filed composition of matter patents (PCTs and
provisionals). We believe our CNS product candidates address
numerous unmet medical needs that can lead to improved efficacy
and/or address tolerability and safety issues that tend to limit
the usefulness of the original approved drug. Our CNS drug
candidates address disease states such as chronic neuropathic pain
(trigeminal neuralgia), myasthenia gravis and vestibular
disorders.
In January 2018, we completed dosing in our Phase 1 clinical trial
for GTP-004, our product candidate for the treatment for the
symptoms of myasthenia gravis. Based on the data, and discussions
with key opinion leaders, we expect to be in a position to initiate
a Phase 2 clinical trial in patients in the second half of
2018.
We also began a proof-of-concept
clinical trial for GTP-011, a 72-hour patch for the prevention of
motion sickness, in late February 2018. We anticipate that the new
drug application, or NDA, will be a 505(b)2 NDA for each of these
programs. PainBrake®
is designed to enable accurate dose fractionation for the treatment
of certain forms of neuropathic pain and we expect to complete the manufacturing tablets for
a bioequivalence study in the second half of 2018 and to conduct a
bioequivalence study subsequently.
CNS Product Candidates
GTP-004
GTP-004
is a fixed-dose combination tablet for the treatment of the muscle
weakness associated with myasthenia gravis, or MG, a chronic
autoimmune disease of the neuromuscular junction characterized by
muscle weakness. MG affects an estimated approximately 36,000 to
60,000 people in the U.S. The basic
abnormality in MG is a reduction in nicotinic acetylcholine
receptors (AChRs) or neighboring proteins at the neuromuscular
junctions (Drachman, 2016). In neonatal myasthenia, the fetus may
acquire antibodies from a mother affected with MG. Generally, cases
of neonatal MG are temporary and the child's symptoms usually
disappear within 2-3 months after birth (Myasthenia Gravis Fact
Sheet; National Institute of neurological Disorders and Stroke,
2016). Rarely, children may have congenital myasthenic syndrome
(CMS) caused by defective gene mutations (Engel, 2012). In some
cases, degeneration of the nerves that innervate muscles such as
occurs with aging (Lexel, 1997) leads to a myasthenic syndrome.
Recently (Makarious et al, 2017), have reported on a myasthenic
syndrome associated with the use of checkpoint
inhibitors.
Cholinesterase
inhibitors, or ChEIs, that do not get into the brain (do not cross
the blood-brain barrier), such as pyridostigmine and neostigmine are used to
treat the muscular weakness associated with myasthenia gravis and
other myasthenic syndromes).
However, ChEIs also act at cholinergic synapses in the gut to cause
GI side effects such as diarrhea, nausea and vomiting, which are
dose-limiting (Engel 2012; Abicht et
al, 2003 updated in 2014).
GTP-004
combines pyridostigmine with ondansetron, designed to attenuate the
gastrointestinal, or GI, side effects of pyridostigmine alone.
Mitigating the GI side effects of pyridostigmine with a drug that
prevents diarrhea, nausea and vomiting should lead to greater
patient comfort, safety, and compliance as well as to improved
efficacy. Several provisional patent applications protecting the
combination of neostigmine or pyridostigmine with a number of
antiemetic drugs were filed by GTP in early 2017.
GTP-004
completed dosing in a Phase 1 clinical trial in January 2018. The
objective of the Phase 1 clinical trial was to demonstrate
that GI side effects of pyridostigmine are reduced with GTP-004.
Healthy volunteers were enrolled in the Phase 1 study.
Following enrollment, subjects received single increasing oral
doses of pyridostigmine (ranging from 30 to 120mg) administered
once daily in the morning. Once subjects experienced intolerable
GI side effects and reached First Intolerable Dose -FID1- as
defined by protocol criteria, upward dose escalation of
pyridostigmine was discontinued and subjects were washed out for 2
to 7 days. Next, subjects that reached FID received daily
increasing doses of pyridostigmine in combination with
ondansetron.
Three
subjects (2 males, one female; aged 34 to 43) reached intolerable
gastrointestinal side effects with pyridostigmine alone. The
dose-limiting gastro-intestinal adverse event occurred at 60 mg for
2 subjects, and 90 mg for the third subject. When these three
subjects received GTP-004 (pyridostigmine with ondansetron),
gastro-intestinal adverse events were abrogated, and all subjects
tolerated doses as high as 120 mg, the maximum allowed dose allowed
by the protocol.
Based
on the data from the Phase 1 clinical trial, and discussions with
key opinion leaders, we expect to be in a position to initiate a
Phase 2 clinical trial in patients in the second half of
2018.
Provisional
patent applications protecting the combination of
Mestinon®
or Prostigmine®
with a number of antiemetic drugs were filed by GTP in early
2017.
PainBrake
PainBrake
is a new patented formulation of carbamazepine
(Tegretol®)
that enables accurate dose fractionation for the treatment of
neuropathic pain, a condition that results from a dysfunction of
nerves involved in the perception of pain and that is typically
chronic and particularly prevalent in elderly patients. An
NIH-supported study published in 2009 estimated that almost 16
million Americans suffer from chronic neuropathic pain (Yawn et
al., 2009) and this number is expected to increase due to the aging
population. Current drugs provide a useful degree of pain relief in
only about half the patients, very few patients achieve complete
relief of pain (Nightingale, 2012). Peak dose-limiting side
effects, mainly sedation, somnolence, dizziness and balance
problems which are poorly tolerated by the elderly (Oomens et al.,
2015) cause patients to be under-dosed, thereby contributing to
inadequate pain relief. This is particularly true for
carbamazepine, a drug that is considered to be the first line
therapy for the treatment of certain forms of neuropathic pain
(Zakrzewska, 2015).
To
overcome dose-limiting side effects, PainBrake tablets employ an
innovative bilayered, deeply scored design patented by AccuBreak.
The top layer contains carbamazepine and is pre-divided by deep
scoring during the manufacturing process to provide accuracy of
dose adjustments by enabling easy tablet splitting into exact
doses. The bottom layer provides mechanical stability and serves as
the break region when splitting the tablet. We have in-licensed against milestones and royalties
the worldwide rights to the use of the AccuBreak technology for the
delivery of drugs that like carbamazepine are voltage-gated sodium
channel blockers. The core patent for the AccuBreak
technology expires in 2025.
PainBrake®
is designed to enable accurate dose fractionation for the treatment
of certain forms of neuropathic pain and we expect to complete the manufacturing tablets for
a bioequivalence study in the second half of 2018 and to begin a
bioequivalence study as a subsequent step.
GTP-011
We are
developing GTP-011 as a 72-hour transdermal product for the
prevention of motion sickness, a well-known syndrome that typically
involves nausea and vomiting in otherwise healthy people and that
occurs upon exposure to certain types of motion.
Currently, the scopolamine patch (Transderm Scop®
from Novartis) is viewed as a
first-line medication for prevention of motion sickness (Gil et
al., 2012; Brainard and Gresham, 2014). However, side effects can
be of particular concern and include sedation (Spinks et al.,
2004), reduced memory for new information, impaired attention, and
lowered feelings of alertness (Parrott, 1989). Mental confusion or
delirium can occur after application of scopolamine patch (Seo et
al., 2009). Elderly people as well as people with undetected
incipient dementia or mild cognitive impairment, or MCI, may be
particularly prone to develop mental confusion after applying the
scopolamine patch (Seo et al., 2009).
GTP-011,
like scopolamine, is a transdermal formulation that contains a
muscarinic receptor antagonist. Unlike scopolamine, however,
GTP-011’s active ingredient has been reported not to affect
memory and cognition and has a low incidence of sedation
(Kay et al., 2012). GTP-011 may
thus be a more favorable alternative, if approved for marketing, to
the scopolamine patch for the treatment of motion sickness. Since
GTP-011 is expected to be a new formulation of an approved drug, we
anticipate that the NDA will be a 505(B)2 NDA. We began a proof-of-concept clinical trial for
GTP-011 in late February 2018.
Our Strategy
Our goal is to be a leader in immuno-oncology therapies targeting a
broad range of indications including hematological malignancies,
sarcoma and solid tumors and to generate value from our CNS product
candidates. Key
elements of our strategy are to:
Expedite clinical development, regulatory approval and
commercialization of our bi-specific ADC, OXS-1550, in specific
indications with a high unmet-medical need such as patients who are
resistant or refractory to conventional treatment and also assess
fast-to-market strategies in potential orphan indications
Based upon promising clinical results from the initial OXS-1550
Phase 1 study, we began enrolling patients in a Phase 2 trial
during the first quarter of 2017 for our most advanced oncology
product candidate, OXS-1550, for the treatment of patients with
relapsed/refractory B-cell leukemias or lymphomas. In the Phase 1
study, of the nine patients who received OXS-1550 at the higher
doses, two had durable complete responses in heavily pretreated
patients. One of these patients, who had failed multiple previous
treatment regimens, has been cancer free since the beginning of
2015.
The approximately 34 patient, open label, two stage, FDA-approved
Phase 2 trial is being conducted at the University of Minnesota's
Masonic Cancer Center. The trial is a continuation of the dose and
schedule finding component of the Phase 1 study using the dose
limiting toxicity identified in the Phase 1 but with a higher
number of cycles. The trial is designed to confirm the safety of
OXS-1550 and make a preliminary determination of activity level by
disease. There
are currently 18 patients enrolled in the Phase 2 trial and
we expect data from this Phase 2 trial
to be available in the second half 2018.
We will also utilize our bi-specific ADC platform to generate novel
ADCs with unique targets, linkers and warheads. We anticipate that
this platform will give us the ability to rapidly construct novel
ADCs with the potential to treat a wide range of cancers, including
hematologic and solid tumors.
Rapidly advanced our Tri-specific Killer Engagers (TriKEs),
OXS-3550 and OXS-C3550
Our
TriKE and TetraKE product candidates have the potential to be
groundbreaking therapies targeting a broad range of hematologic
malignancies, sarcomas and solid tumors. We are preparing to study
OXS-3550, an anti-CD16-IL-15-anti-CD33 TriKE in CD33 positive
leukemias, a marker expressed on tumor cells in AML, MDS and other
myeloid malignancies. We expect to begin a Phase 1 clinical trial
in the second half of 2018 in patients with relapsed/refractory
AML. The Phase 1 trial will be a dose finding study. We expect this
will be closely followed by Phase 2 trials to determine the most
efficacious dosing and cycles with the aim to maximize efficacy
while minimizing on-target, off-disease adverse
events.
OXS-C3550 contains a humanized single-domain anti-CD16 moiety which
demonstrated improved binding characteristics and enhanced tumor
cell killing based on functional assays and animal models of
AML.
We have
designed OXS-3550 and OXS-C3550, if approved for marketing, to
serve as a relatively safe, cost-effective, and easy-to-use
therapies for resistant/relapsing AML or MDS which could also be
combined with chemotherapy as frontline therapy thus targeting a
broad AML/MDS market.
OXS-C3550 is a next-generation, follow-on, to our lead TriKE,
OXS-3550. OXS-3550 studies will help inform the development of
OXS-C3550. We believe this will de-risk the OXS-C3550 program as
the data being generated will help to make informed decisions on
which, or both, will be brought into later phase studies and in
which patient populations.
Utilize our TriKE and TetraKE platform technologies to develop a
robust pipeline of targeted immuno-oncology products targeting a
wide range of hematologic malignancies, sarcomas and solid tumors
for development on our own and through potential collaborations
with larger pharmaceutical companies
We are using our TriKE and TetraKE platforms with the intent to
bring to market multiple, targeted, off-the-shelf therapies that
can treat a range of hematologic malignancies, sarcomas and solid
tumors. The platforms are scalable and we are currently working
with several third parties investigating the optimal expression
system of the TriKEs and TetraKE constructs which we expect to be
part of a process in which we are able to produce IND-ready
moieties in approximately 90-120 days after the construct
conceptual design. After conducting market and competitive
research, specific moieties can then be rapidly advanced into the
clinic on our own or through potential collaborations with larger
pharmaceutical companies.
We are currently evaluating over a dozen moieties and intend to
announce additional clinical product candidates in the second half
of 2018. We intend to initiate human clinical testing for certain
of our solid tumor product candidates in 2019.
We believe our TriKEs and TetraKEs will have the ability, if
approved for marketing, to be used on a stand-alone basis, augment
the current monoclonal antibody therapeutics, or be used in
conjunction with more traditional cancer therapy and potentially
overcome certain limitations of current chimeric antigen receptor,
or CAR-T, therapy.
Continue our collaborative relationship with the Masonic Cancer
Center at the University of Minnesota, under a program led by Dr.
Jeffrey Miller and become the leading NK-oriented
immune-oncology company
We believe that the TriKE and TetraKE constructs represent
potentially groundbreaking innovations in immunotherapy. In July
2016 we entered into an exclusive license agreement with the
University of Minnesota to
develop and commercialize cancer therapies using TriKE and TetraKE
technology developed by researchers at the university to target NK
cells to cancer.
We believe TriKE and TetraKE therapeutics have the potential to
significantly impact the standard of care for hematologic
malignancies, sarcomas, as well as solid tumors. The direct
engagement of the NK cell with the tumor cell via very specific
receptors may increase the efficacy while decrease the toxicity
seen with other forms of immunotherapies. If approved, we expect
the TriKEs and TetraKEs will be able to be administered at cancer
treatment facilities without the need for specialized centers or
product-specific trained staff.
We also intend to selectively evaluate and potentially acquire or
enter into licensing or other agreements for technologies and/or
product candidates that we believe would complement our oncology
product candidates and platform technologies.
Monetize our CNS programs through transactions with
commercialization-oriented pharmaceutical companies and/or other
transactions
Our CNS portfolio consists of innovative reformulations and/or
repurposing of existing therapies and are covered by formulation
patents or filed composition of matter patents (PCTs and
provisional applications) and represent, what we believe to be,
near-to-market product opportunities. Our CNS programs address
numerous unmet medical needs that, if approved, we believe may lead
to improved efficacy while addressing tolerability and safety
issues that tend to limit the usefulness of the original approved
drugs.
We expect to take advantage of our CNS portfolio by generating
proof-of-concept data and/or achieving other milestones and
ultimately entering into transactions with
commercialization-oriented pharmaceutical companies, which could
result in income, or enter into other transaction structures with
the intent to generate value for our shareholders.
Oncology Markets
B-cell Lymphomas/Leukemias
B-cell
lymphoma is a type of cancer that forms in B cells (a type of
immune system cell). Bcell lymphomas may be either indolent
(slow-growing) or aggressive (fast-growing). Most Bcell
lymphomas are non-Hodgkin lymphomas. There are many different types
of B-cell non-Hodgkin lymphomas. These include Burkitt lymphoma,
chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL),
diffuse large B-cell lymphoma, follicular lymphoma, and mantle cell
lymphoma. It is the most common type of non- Hodgkin lymphoma among
adults, with an annual incidence of 7–8 cases per 100,000
people per year.
Acute Lymphoblastic Leukemia
Acute
lymphoblastic leukemia, or ALL, is an acute form of leukemia, or cancer of the
white blood cells, characterized by the overproduction and
accumulation of immature white blood cells, known as lymphoblasts.
In persons with ALL, lymphoblasts are overproduced in the bone
marrow and continuously multiply, causing damage and death by
inhibiting the production of normal cells (such as red and white
blood cells and platelets) in the bone marrow and by spreading
(infiltrating) to other organs.
It is
estimated that there will be approximately 6,000 new cases of ALL
reported in the United States in 2018 (ACS Cancer Facts &
Figures 2018). "Acute" is defined by the World Health Organization
standards, in which greater than 20% of the cells in the bone
marrow are blasts. Chronic lymphocytic leukemia is defined as
having less than 20% blasts in the bone marrow. Acute lymphoblastic
leukemia is seen in both children and adults; the highest incidence
is seen between ages 2 and 5 years. ALL is the most common
childhood cancer constituting about 23 to 30% of cancers before age
15. Although 80 to 90% of children will have a durable complete
response with treatment it is the leading cause of cancer-related
deaths among children.
Multiple Myeloma
Multiple
myeloma is a type of cancer that forms in white blood cells and
will affect an estimated 30,770 people in 2018 in the U.S. causing
about 12,770 deaths. Multiple myeloma causes cancer cells to
accumulate in the bone marrow, where they crowd out healthy blood
cells. Multiple myeloma is also characterized by destructive lytic
bone lesions (rounded, punched-out areas of bone), diffuse
osteoporosis, bone pain, and the production of abnormal proteins
which accumulate in the urine. Anemia is also present in most
multiple myeloma patients at the time of diagnosis and during
follow-up. Anemia in multiple myeloma is multifactorial and is
secondary to bone marrow replacement by malignant plasma cells,
chronic inflammation, relative erythropoietin deficiency, and
vitamin deficiency. Plasma cell leukemia, a condition in which
plasma cells comprise greater than 20% of peripheral leukocytes, is
typically a terminal stage of multiple myeloma and is associated
with short survival.
Myeloid Leukemias
Acute Myeloid Leukemia
AML is a heterogeneous hematologic stem cell malignancy in adults
with incidence rate of 3–5% per 100,000 populations. The
median age at the time of diagnosis is 65–69 years. AML is an
aggressive disease and is fatal without anti-leukemic treatment.
AML is the most common form of adult leukemia with 20,000 new cases
each year. These patients will require frontline therapy, usually
chemotherapy including cytarabine and an anthracycline, a therapy
that has not changed in over 40 years. Myelodysplastic syndromes
(MDS) are a heterogeneous group of myeloid neoplasms characterized
by dysplastic features of erythroid/myeloid/megakaryocytic
lineages, progressive bone marrow failure, a varying percentage of
blast cells, and enhanced risk to evolve into acute myeloid
leukemia. It is estimated that over 10,000 new cases of MDS are
diagnosed each year and there are minimal treatment options; other
estimates have put this number higher. In addition, the incidence
of MDS is rising for unknown reasons.
Solid Tumors
In the
United States, in 2018, it is estimated there will be approximately
1,735,350 new cases of cancer resulting in over 600,000 deaths.
Greater than 80% of these cancers will be classified as solid
tumors. The most prevalent new cases of solid tumors being breast,
lung, prostate, colorectal and bladder. (American Cancer Society,
Cancer Facts & Figures 2018)
Sarcomas
A
sarcoma is a type of cancer that develops from certain tissues,
like bone or muscle. Bone and soft tissue sarcomas are the main
types of sarcoma. Soft tissue sarcomas can develop from soft
tissues like fat, muscle, nerves, fibrous tissues, blood vessels,
or deep skin tissues. They can be found in any part of the body.
Most of them develop in the arms or legs. They can also be found in
the trunk, head and neck area, internal organs, and the area in
back of the abdominal cavity (known as the retroperitoneum).
Sarcomas are not common tumors, and most cancers are the type of
tumors called carcinomas.
The
American Cancer Society's estimates for soft tissue sarcomas in the
United States for 2018 are (these statistics include both adults
and children): about 13,040 new soft tissue sarcomas will be
diagnosed (7,370 cases in males and 5,670 cases in females). 5,150
Americans (2,770 males and 2,380 females) are expected to die of
soft tissue sarcomas. The most common types of sarcoma in adults
are undifferentiated pleomorphic sarcoma (previously called
malignant fibrous histiocytoma), liposarcoma, and leiomyosarcoma.
Certain types occur more often in certain areas of the body than
others. For example, leiomyosarcomas are the most common abdominal
sarcoma, while liposarcomas and undifferentiated pleomorphic
sarcoma are most common in legs. But pathologists (doctors who
specialize in diagnosing cancers by how they look under the
microscope), may not always agree on the exact type of sarcoma.
Sarcomas of uncertain type are very common. (American Cancer
Society, Cancer Facts & Figures 2018)
CNS Markets
Chronic Neuropathic Pain
Neuropathic
pain has many causes, including trigeminal neuralgia, diabetes,
certain forms of chemotherapy, trauma, toxins, infection such as
postherpetic infection, immune deficiencies, ischemic disorders,
and multiple sclerosis. According to the latest market report
published by Persistence Market Research (November 2016) titled
“Global Market Study on Neuropathic Pain:
Anticonvulsants Drug Class Segment Projected to Witness the Highest
Growth Through 2024,”
the global neuropathic pain market was valued
at $5.2 billion in 2015 and was estimated to
reach a market valuation of $5.4 billion by 2016. The market is projected
to expand at a compound annual growth rate of 5.6% during an
eight-year forecast period 2016–2024 and
reach $8.3 billion by the end of
2024.
The chronic pain market continues to represent a major unmet
medical need. Current drugs provide a useful degree of pain relief
in only about half the patients (Nightingale, 2012). It is
estimated that only one in four patients with neuropathic pain
experiences over 50% pain relief, and 30% of patients have no or
very little relief. Very few patients achieve complete pain relief.
In most patients, pain relief is obtained at the price of
burdensome side effects. For many drugs, inadequate pain relief is
due to side effects that occur when drugs reach peak concentrations
in the blood, these side effects are dose-dependent and
dose-limiting and prevent the use of fully effective doses. As a
consequence, patients are chronically under-dosed. This is
particularly true for carbamazepine, a drug with which it may be
possible to achieve nearly complete relief of pain in many patients
who suffer from forms of neuropathic pain that respond to
carbamazepine.
Current
treatments for neuropathic pain treat the symptom (pain) and
include narcotic analgesics, voltage-gated sodium channel blockers,
voltage-gated calcium channel blockers, glutamate NMDA NR2B
antagonists (ketamine), drugs that increase monoamine transmission,
and cannabinoids. However, these therapies have safety and
tolerability issues including, for some, tolerance, abuse and
addiction liability. Many have dose-limiting side effects that
prevent patients from receiving fully effective doses of
medication, further limiting efficacy. Some of the key players operating in the global
neuropathic pain market are Depomed Inc. (NASDAQ:DEPO), Pfizer Inc.
(NYSE:PFE), Johnson & Johnson (NYSE:JNJ), Bristol-Myers Squibb
(NYSE:BMY), Eli Lilly and Company (NYSE:LLY), GlaxoSmithKline PLC
(NYSE:GSK), Sanofi S.A. (NYSE:SNY), Biogen Idec Inc. (NASDAQ:BIIB),
and Baxter Healthcare Corporation (NYSE:BAX), among
others.
Myasthenia Gravis
MG is a
rare, chronic autoimmune disease of the neuromuscular junction
caused by antibodies that attack components of the postsynaptic
membrane, impair neuromuscular transmission, and lead to varying
degrees of weakness and fatigue of skeletal muscle. Recently,
anti-cancer treatments with check-point inhibitors have been
associated in some patients with rapidly progressive severe
myasthenia gravis. The prevalence of MG in the United States is
estimated at 14 to 20 per 100,000 population, approximately 36,000
to 60,000 cases in the US (Howard, 2015). Rarely, children may show signs of congenital
myasthenia or congenital myasthenic syndrome (CMS). These are not
autoimmune disorders, but are caused by defective genes that
produce abnormal proteins instead of those that normally are
involved in cholinergic transmission: acetylcholinesterase (the
enzyme that breaks down acetylcholine), acetylcholine receptors,
and other proteins present along the muscle membrane (Engel,
2012).In some rare cases, a myasthenic syndrome is due to
bi-allelic variants in the gene encoding the vesicular
acetylcholine transporter (VAChT) located in the presynaptic
terminal (O’Grady et al, 2016). In other cases, degeneration
of the nerves that innervate muscles such as occurs with aging
(Lexel, 1997) leads to a myasthenic syndrome.
Only
two drugs are currently approved for the muscular symptoms of
myasthenia gravis, namely Mestinon® and
Prostigmine®. Both have the
same mechanism of action, and both are associated with
gastrointestinal side effects, which are an important source of discomfort for the
patient, may be the source of non-compliance, or may result in the
need to decrease the dose of ChEI to mitigate these side effects
when these become dose-limiting. In many patients the side effects
are dose limiting and prevent the administration of the fully
effective dose. Attempts at overcoming these side effects using
dose fractionation or a slow release formulation (Mestinon
TimeSpan) have been disappointing.
Motion Sickness
The current transdermal market leader for the prevention of motion
sickness is the 72-hour scopolamine patch (Gil et al., 2012)
commercialized by Sandoz. However, this medication can cause a
number of worrisome side effects., especially in the elderly, such
as confusion and memory impairment. The scopolamine patch is not
approved for children.
Manufacturing
We do not currently own or operate manufacturing facilities for the
production of clinical or commercial quantities of any of our
product candidates. We rely on a small number of third-party
manufacturers to produce our compounds and expect to continue to do
so to meet the preclinical and clinical requirements of our
potential product candidates as well as for all of our commercial
needs. We do not have long-term agreements with any of these third
parties. We require in our manufacturing and processing agreements
that all third-party contract manufacturers and processors produce
active pharmaceutical ingredients, or API, and finished products in
accordance with the FDA’s current Good Manufacturing
Practices, or cGMP, and all other applicable laws and regulations.
We maintain confidentiality agreements with potential and existing
manufacturers in order to protect our proprietary rights related to
our drug candidates.
Patents and Trademarks
University of Minnesota License Agreement
We
(through our wholly owned subsidiary Oxis Biotech, Inc.) are party
to an exclusive worldwide license agreement with the Regents of the
University of Minnesota, to further develop and commercialize
cancer therapies using TriKE technology developed by researchers at
the university to target NK cells to cancer. Under the terms of the
agreement, we receive exclusive rights to conduct research and to
develop, make, use, sell, and import TriKE technology worldwide for
the treatment of any disease, state or condition in humans. We
shall be responsible for obtaining all permits, licenses,
authorizations, registrations and regulatory approvals required or
granted by any governmental authority anywhere in the world that is
responsible for the regulation of products such as the TriKE
technology, including without limitation the FDA in the United
States and the European Agency for the Evaluation of Medicinal
Products in the European Union. Under the agreement, the University
of Minnesota will receive an upfront license fee, royalty fees
ranging from 4% to 6%, minimum annual royalty payments of $250,000
beginning in 2022, $2,000,000 in 2025, and $5,000,000 in 2027 and
certain milestone payments totaling $3,100,000.
The
following is a list of the patent applications that we licensed
from the University of Minnesota:
|
Appl. No.
|
Title
|
Country
|
Status
|
|
U.S.
Patent Application Number 62/237,835
|
Therapeutic
compounds and its uses
|
US
|
Expired
|
|
PCT
Patent Application Number PCT/US2016/055722
|
Therapeutic
compounds and methods
|
US
|
Pending
|
Daniel A. Vallera, Ph.D. License Agreement
We are
party to an exclusive worldwide license agreement with Daniel A.
Vallera, Ph.D. and his co-inventor Jeffrey Lion, or jointly, Dr.
Vallera, to further develop and commercialize DT2219ARL
(OXS1550), a novel therapy for the treatment of various human
cancers. Under the terms of the agreement, we receive exclusive
rights to conduct research and to develop, make, use, sell, and
import DT2219ARL worldwide for the treatment of any disease, state
or condition in humans. We shall be responsible for obtaining all
permits, licenses, authorizations, registrations and regulatory
approvals required or granted by any governmental authority
anywhere in the world that is responsible for the regulation of
products such as DT2219ARL, including without limitation the FDA in
the United States and the European Agency for the Evaluation of
Medicinal Products in the European Union. Under the agreement, Dr.
Vallera will receive an upfront license fee, royalty fees ranging
from 3% for net sales and 25% of net sublicensing revenues, and
certain milestone payments totaling $1,500,000.
The
following is a list of the patent applications and patents that we
licensed from Dr. Vallera under our license
agreements:
|
Pat./Pub. No.
|
Title
|
Country
|
Status
|
|
U.S.
Patent Application Number 61/160,530
|
Methods
and compositions for bi-specific targeting of
cd19/cd22
|
US
|
Expired
|
|
U.S.
Patent Number 9,371,386
|
Methods
and compositions for bi-specific targeting of
cd19/cd22
|
US
|
Issued
|
|
U.S.
Patent Application Number 15/187,579
|
Methods
and compositions for bi-specific targeting of
cd19/cd22
|
US
|
Pending
|
ID4 License Agreement
Pursuant
to a patent license agreement with ID4, dated December 31, 2014, or
the ID4 License Agreement, we received an exclusive, worldwide
license to certain intellectual property, including intellectual
property related to treating a p62mediated disease (e.g.,
multiple myeloma). The terms of this license require us to pay ID4
royalties equal to 3% of net sales of products and 25% royalty of
net sublicensing revenues. The license will expire upon expiration
of the last patent contained in the licensed patent rights, unless
terminated earlier. We may terminate the licensing agreement with
ID4 by providing ID4 with a 30 days written notice.
We will
owe the following cash amounts to ID4 Pharma upon the attainment of
the following milestones:
(i)
Filing of an
investigational new drug application with a competent regulatory
authority anywhere in the world $50,000.
(ii)
Initiation of Phase
I Human Clinical Trial: $50,000.
(iii)
Initiation of Phase
II Human Clinical Trial: $100,000.
(iv)
Initiation of
pivotal Phase III Human Clinical Trial: $250,000. and
(v)
Receipt of the
first marketing approval: $250,000
The
following is a list of the patent applications and patent that we
licensed from ID4 under the ID4 license agreement:
|
Pat./Appl. No.
|
Title
|
Country
|
Status
|
|
U.S.
Patent Number
9,580,382
|
P62zz
chemical inhibitor
|
US
|
Issued
|
|
U.S.
Patent Application Number 61/521,287
|
P62zz
chemical inhibitor
|
US
|
Expired
|
|
PCT
Patent Application Number PCT/US2012/049911
|
P62zz
chemical inhibitor
|
PCT
|
Expired
|
|
U.S.
Patent Application Number 14/727,710
|
P62zz
chemical inhibitor
|
US
|
Pending
|
|
Chinese
Patent Application 201280048718
|
P62zz
chemical inhibitor
|
US
|
Pending
|
Patents for AccuBreak Tablets
We have
in-licensed the rights to use the AccuBreak patents with drugs
that, like carbamazepine, are voltage-gated sodium channel blockers
in North America. The license field includes voltage gated sodium
channels inhibitors and blockers for the treatment of epilepsy,
neuropathic pain, and bipolar disorder.
Under
the agreement, AccuBreak received an upfront license fee of
$35,000, royalty fees ranging from 2.5% to 5%, minimum annual
royalty payments, and 20% of net sublicensing
revenues.
We will
owe the following cash amounts to AccuBreak upon the attainment of
the following milestones:
●
$50,000 six months
after the first approval of the first indication by the
FDA;
●
$50,000 nine months
after the first approval of the first indication by the
FDA;
●
$100,000 12 months
after the first approval of the first indication by the
FDA;
●
$25,000 upon
achievement of $25,000,000 of cumulative net sales in the
world;
●
$50,000 upon
achievement of $50,000,000 of cumulative net sales in the world;
and
●
$100,000 upon
achievement of $75,000,000 of cumulative net sales in the
world.
Four formulation patents protect the AccuBreak
Technology:
|
Pat. No.
|
Title
|
Country
|
Status
|
|
U.S.
Patent Number 7,838,031
|
Method for administering a partial dose using a segmented
pharmaceutical tablet
|
US
|
Issued
|
|
U.S.
Patent Number 7,879,352
|
Scored pharmaceutical tablets comprising a plurality of
segments
|
US
|
Issued
|
|
U.S.
Patent Number 8,158,148
|
Pharmaceutical tablets comprising two or more unitary
segments
|
US
|
Issued
|
|
U.S.
Patent Number 8,231,902 (ABT-054)
|
Segmented pharmaceutical dosage forms
|
US
|
Issued
|
The core patent expires in 2025.
Patent Applications for GTP-004
Four patent applications filed by GTP in 2017 with the U.S. PTO
protect the combination of pyridostigmine or neostigmine + an
antiemetic for the treatment of myasthenia gravis. We plan to file
extensions under the Patent Cooperation Treaty, or PCT, in 2018.
All patents list below are owned by the Company.
|
Pat. No.
|
Title
|
Country
|
Status
|
|
U.S.
Patent Application Number 62/443,904
|
Use and composition for treating Myasthenia Gravis
|
US
|
Expired
|
|
U.S.
Patent Application Number 62/449,699
|
Neostigmine combination for treating Myasthenia Gravis
|
US
|
Expired
|
|
U.S.
Patent Application Number 62/536,595
|
Method and composition for treating Myasthenia Gravis
|
US
|
Pending
|
|
U.S.
Patent Application Number 62/536,580
|
Neostigmine pharmaceutical combination for treating Myasthenia
Gravis
|
US
|
Pending
|
|
PCT
Application Number
PCT/US/18/12754
|
Use and
composition for treating Myasthenia Gravis
|
PCT
|
Claims
priority from US 62/443,904
|
|
Taiwan
Application Number
107100813
|
|
TW
|
Awaiting
FC Report
|
|
PCT
Application Number
PCT/US18/014700
|
Neostigmine
pharmaceutical combination for treating Myasthenia
Gravis
|
PCT
|
Claims
priority from US 62/449,699
|
|
Taiwan
Application Number
101702591
|
|
TW
|
Awaiting
FC Report
|
Patent Application for GTP-011
One
patent application filed by GTP in 2017 with the U.S. PTO protects
a 72-hour patch of oxybutynin for the treatment of motion sickness.
We plan to file a PCT extension in 2018. All patents list below are owned by the
Company.
|
Appl. No.
|
Title
|
Country
|
Status
|
|
U.S.
Patent Application Number 62/440,575
|
Use and composition for preventing and treating motion
sickness
|
US
|
Expired
|
|
US
Patent Application
Number
62/595,667
|
Use,
method, and device for the prevention and treatment of motion
sickness
|
US
|
Pending*
|
|
PCT
Application Number
PCT/US/17/68944
|
Use and
composition for preventing and treating motion
sickness
|
PCT
|
Claims
priority from US
62/440,575
|
|
Taiwan
Application Number
107100079
|
|
TW
|
Awaiting
FC Report
|
|
* This
application is pending, but was used as priority document of the
PCT ‘944, including its subject matter
|
Research and Development
Expenditures
for research and development activities related to continuing
operations were $1,068,000 and $975,000 million for the years ended
December 31, 2017 and 2016, respectively.
Our
currently projected expenditures for 2018 include approximately $10
million to $12 million for research and development. The actual
cost of our programs could differ significantly from our current
projections if we change our planned development process. In the
event that actual costs of our clinical program, or any of our
other ongoing research activities, are significantly higher than
our current estimates, we may be required to significantly modify
our planned level of operations.
The
successful development of any product candidate is highly
uncertain. It is difficult to reasonably estimate or know the
nature, timing and costs of the efforts necessary to complete the
development of, or the period in which material net cash inflows
are expected to commence from any product candidate, due to the
numerous risks and uncertainties associated with developing drugs.
Any failure to complete any stage of the development of products in
a timely manner could have a material adverse effect on our
operations, financial position and liquidity.
Competition
The
biotechnology and pharmaceutical industries are subject to rapid
technological change. Competition from domestic and foreign
biotechnology companies, large pharmaceutical companies and other
institutions is intense and expected to increase. A number of
companies are pursuing the development of pharmaceuticals in our
targeted areas. According to a recent analysis by InVentiv Health
there are over 800 companies developing approximately 1500 cancer
immunotherapies via 4000 development projects across 535 targets.
According to the Pharmaceutical Manufacturers Research Association,
at the end of 2015 there were 135 drugs in development for the
treatment of lymphomas (blood cell cancers including multiple
myeloma).
Many
compounds are in development for the treatment of neuropathic pain.
Current treatments for neuropathic include narcotic analgesics,
voltage-gated sodium channel blockers, voltage-gated calcium
channel blockers, glutamate NMDA NR2B antagonists (ketamine), drugs
that increase monoamine transmission, and cannabinoids.
Some of the key players operating in
the global neuropathic pain market are Depomed Inc. (NASDAQ:DEPO),
Pfizer Inc. (NYSE:PFE), Johnson & Johnson (NYSE:JNJ),
Bristol-Myers Squibb (NYSE:BMY), Eli Lilly and Company (NYSE:LLY),
GlaxoSmithKline PLC (NYSE:GSK), Sanofi S.A. (NYSE:SNY), Biogen Idec
Inc. (NASDAQ:BIIB), and Baxter Healthcare Corporation (NYSE:BAX),
among others. In the field of myasthenia gravis, pharmaceutical
research and development efforts focus on the discovery of a cure
for the disease. A cure would make treatment with GTP-004 obsolete.
In the field of motion sickness, research may be ongoing for better
anti-motion sickness drugs.
Government Regulation
Government Regulation and Product Approval
Government
authorities in the United States at the federal, state and local
level, and in other countries, extensively regulate, among other
things, the research, development, testing, manufacture, (including
manufacturing changes), quality control, approval, labeling,
packaging, storage, record-keeping, promotion, advertising,
distribution, marketing, export and import of products such as
those we are developing. The processes for obtaining regulatory
approvals in the United States and in foreign countries, along with
subsequent compliance with applicable statutes and regulations,
require the expenditure of substantial time and financial
resources.
U.S. Drug Development Process
In the
United States, the FDA regulates drugs and biological products
under the FDCA and the FDA's implementing regulations.
Failure
to comply with the applicable U.S. requirements at any time during
the product development process, approval process, or after
approval, may subject an applicant to administrative or judicial
sanctions. These sanctions could include the FDA's refusal to
approve pending applications, withdrawal of an approval, a clinical
hold, untitled or warning letters, product recalls, product
seizures, total or partial suspension of production or
distribution, injunctions, fines, refusals of government contracts,
restitution, disgorgement or civil or criminal penalties. The
process required by the FDA before a drug or biologic may be
marketed in the United States generally involves the
following:
●
completion of
preclinical laboratory tests, animal studies and formulation
studies according to Good Laboratory Practices
regulations;
●
submission to the
FDA of an IND, which must become effective before human clinical
studies may begin;
●
approval by an
independent IRB, at each clinical site before each trial may be
initiated;
●
performance of
adequate and well-controlled human clinical studies according to
GCP regulations, to establish the safety and efficacy of the
proposed drug or biologic for its intended use;
●
preparation and
submission to the FDA of an NDA or BLA;
●
satisfactory
completion of an FDA inspection of the manufacturing facility or
facilities at which the product, or components thereof, are
produced to assess compliance with cGMP to assure that the
facilities, methods, and controls are adequate to preserve the
drug's or biologic’s identity, strength, quality, and purity;
and
●
FDA review and
approval of the NDA or BLA.
The
testing and approval process requires substantial time, effort and
financial resources, and we cannot be certain that any approvals
for our product candidates (or those of our collaborators or
licensees) will be granted on a timely basis, if at
all.
An IND
sponsor must submit the results of the preclinical tests, together
with manufacturing information, analytical data and any available
clinical data or literature, to the FDA as part of the IND. The
sponsor must also include a protocol detailing, among other things,
the objectives of the initial clinical study, the parameters to be
used in monitoring safety and the effectiveness criteria to be
evaluated if the initial clinical study lends itself to an efficacy
evaluation. Some preclinical testing may continue even after the
IND is submitted. The IND automatically becomes effective
30 days after receipt by the FDA, unless the FDA raises
concerns or questions related to a proposed clinical study and
places the study on a clinical hold within that 30-day time period.
In such a case, the IND sponsor and the FDA must resolve any
outstanding concerns before the clinical study can begin. Clinical
holds also may be imposed by the FDA at any time before or during
clinical studies due to safety concerns or non-compliance, and may
be imposed on all product candidates within a certain
pharmaceutical class. The FDA also can impose partial clinical
holds, for example, prohibiting the initiation of clinical studies
of a certain duration or for a certain dose.
All
clinical studies must be conducted under the supervision of one or
more qualified investigators in accordance with GCP regulations.
These regulations include the requirement that all research
subjects provide informed consent in writing before their
participation in any clinical study. Further, an IRB must review
and approve the plan for any clinical study before it commences at
any institution, and the IRB must conduct continuing review and
reapprove the study at least annually. An IRB considers, among
other things, whether the risks to individuals participating in the
clinical study are minimized and are reasonable in relation to
anticipated benefits. The IRB also approves the information
regarding the clinical study and the consent form that must be
provided to each clinical study subject or his or her legal
representative and must monitor the clinical study until completed.
Each new clinical protocol and any amendments to the protocol must
be submitted for FDA review, and to the IRBs for approval.
Protocols detail, among other things, the objectives of the
clinical study, dosing procedures, subject selection and exclusion
criteria, and the parameters to be used to monitor subject safety.
Study sites are subject to inspection for compliance with
GCP.
Information
about certain clinical trials must be submitted within specific
timeframes to the National Institutes of Health (NIH), for public
dissemination on their ClinicalTrials.gov website.
Human
clinical studies are typically conducted in three sequential phases
that may overlap or be combined:
●
Phase 1. The product is
initially introduced into a small number of healthy human subjects
or patients and tested for safety, dosage tolerance, absorption,
metabolism, distribution and excretion and, if possible, to gain
early evidence on effectiveness. In the case of some products for
severe or life-threatening diseases, especially when the product is
suspected or known to be unavoidably toxic, the initial human
testing may be conducted in patients.
●
Phase 2. Involves
clinical studies in a limited patient population to identify
possible adverse effects and safety risks, to preliminarily
evaluate the efficacy of the product for specific targeted diseases
and to determine dosage tolerance and optimal dosage and
schedule.
●
Phase 3. Clinical
studies are undertaken to further evaluate dosage, clinical
efficacy and safety in an expanded patient population at
geographically dispersed clinical study sites. These clinical
studies are intended to establish the overall risk/benefit
relationship of the product and provide an adequate basis for
product labeling.
Progress
reports detailing the results of the clinical studies must be
submitted at least annually to the FDA and safety reports must be
submitted to the FDA and the investigators for serious and
unexpected suspected adverse events. Phase 1, Phase 2 and
Phase 3 testing may not be completed successfully within any
specified period, if at all. The FDA or the sponsor may suspend or
terminate a clinical study at any time on various grounds,
including a finding that the research subjects or patients are
being exposed to an unacceptable health risk. Similarly, an IRB can
suspend or terminate approval of a clinical study at its
institution if the clinical study is not being conducted in
accordance with the IRB's requirements or if the drug has been
associated with unexpected serious harm to patients.
U.S. Review and Approval Processes
Assuming
successful completion of the required clinical testing, the results
of product development, preclinical studies and clinical studies,
along with descriptions of the manufacturing process, analytical
tests conducted on the drug, proposed labeling and other relevant
information, are submitted to the FDA as part of an NDA for a new
drug or BLA for a new biological drug product, requesting approval
to market the product.
The
submission of an NDA or BLA is subject to the payment of a
substantial application user fee although a waiver of such fee may
be obtained under certain limited circumstances. For example, the
agency will waive the application fee for an orphan product or for
the first human drug application that a small business or its
affiliate submits for review. The sponsor of an approved NDA or BLA
is also subject to annual product and establishment user
fees
In
addition, under the Pediatric Research Equity Act of 2003, or PREA,
an NDA or BLA application (or supplements to an application) for a
new active ingredient, new indication, new dosage form, new dosing
regimen, or new route of administration must contain data that are
adequate to assess the safety and effectiveness of the drug or
biologic for the claimed indications in all relevant pediatric
subpopulations, and to support dosing and administration for each
pediatric subpopulation for which the product is safe and
effective, unless the applicant has obtained a waiver or
deferral.
In
2012, the FDASIA amended the FDCA to require that a sponsor who is
planning to submit a marketing application for a drug that includes
a new active ingredient, new indication, new dosage form, new
dosing regimen or new route of administration submit an initial
Pediatric Study Plan, or PSP, within sixty days of an
End-of-Phase 2 meeting or as may be agreed between the sponsor
and the FDA. The initial PSP must include an outline of the
pediatric study or studies that the sponsor plans to conduct,
including study objectives and design, age groups, relevant
endpoints and statistical approach, or a justification for not
including such detailed information, and any request for a deferral
of pediatric assessments or a full or partial waiver of the
requirement to provide data from pediatric studies along with
supporting information. The FDA may, on its own initiative or at
the request of the applicant, grant deferrals for submission of
data or full or partial waivers. The FDA and the sponsor must reach
agreement on the PSP. A sponsor can submit amendments to an
agreed-upon initial PSP at any time if changes to the pediatric
plan need to be considered based on data collected from preclinical
studies, early phase clinical studies, and/or other clinical
development programs.
The FDA
also may require submission of a REMS to mitigate any identified or
suspected serious risks. The REMS could include medication guides,
physician communication plans, assessment plans, and elements to
assure safe use, such as restricted distribution methods, patient
registries, or other risk minimization tools.
The FDA
reviews all NDAs and BLAs submitted to ensure that they are
sufficiently complete for substantive review before it accepts them
for filing. The FDA may request additional information rather than
accept an application for filing. In this event, the application
must be re-submitted with the additional information. The
re-submitted application also is subject to review before the FDA
accepts it for filing. Once the submission is accepted for filing,
the FDA begins an in-depth substantive review to determine whether
the product is safe and effective for its intended
use.
The FDA
may refer the NDA or BLA to an advisory committee for review,
evaluation and recommendation as to whether the application should
be approved and under what conditions. An advisory committee is a
panel of experts, including clinicians and other scientific
experts, who provide advice and recommendations when requested by
the FDA. The FDA is not bound by the recommendation of an advisory
committee, but it considers such recommendations when making
decisions.
Additionally,
before approving an NDA or BLA, the FDA will typically inspect one
or more clinical sites to assure clinical data supporting the
submission were developed in compliance with GCP.
The
approval process is lengthy and difficult and the FDA may refuse to
approve an NDA or BLA if the applicable regulatory criteria are not
satisfied, or may require additional clinical data or other data
and information. Even if such data and information are submitted,
the FDA may ultimately decide that the NDA or BLA does not satisfy
the criteria for approval. Data obtained from clinical studies are
not always conclusive and the FDA may interpret data differently
than an applicant interprets the same data.
After
the FDA's evaluation of an application, the FDA may issue an
approval letter, or, in some cases, a complete response letter to
indicate that the review cycle is complete and that the application
is not ready for approval. A complete response letter generally
contains a statement of specific conditions that must be met to
secure final approval of the application and may require additional
clinical or preclinical testing for the FDA to reconsider the
application. The deficiencies identified may be minor, for example,
requiring labeling changes, or major, for example, requiring
additional clinical studies. Additionally, the complete response
letter may include recommended actions that the applicant might
take to place the application in a condition for approval. If a
complete response letter is issued, the applicant may either
resubmit the application, addressing all of the deficiencies
identified in the letter, or withdraw the application or request an
opportunity for a hearing.
Even
with submission of additional information, the FDA ultimately may
decide that the application does not satisfy the regulatory
criteria for approval. If and when those conditions have been met
to the FDA's satisfaction, the FDA will typically issue an approval
letter. An approval letter authorizes commercial marketing of the
drug with specific prescribing information for specific
indications.
If a
product receives regulatory approval, the approval may be
significantly limited to specific diseases and dosages or the
indications for use may otherwise be limited, which could restrict
the commercial value of the product. Further, the FDA may require
that certain contraindications, warnings or precautions be included
in the product labeling. In addition, the FDA may require
post-approval studies, including Phase 4 clinical studies, to
further assess safety and effectiveness after approval and may
require testing and surveillance programs to monitor the safety of
approved products that have been commercialized. After approval,
some types of changes to the approved product, such as adding new
indications, manufacturing changes, and additional labeling claims,
are subject to further testing requirements and FDA review and
approval.
ANDAs and Section 505(b)(2) New Drug Applications
Most
drug products obtain FDA marketing approval pursuant to an NDA or
BLA (described above) for innovator products, or an ANDA for
generic products. Relevant to ANDAs, the Hatch-Waxman amendments to
the FDCA established a statutory procedure for submission and FDA
review and approval of ANDAs for generic versions of branded drugs
previously approved by the FDA (such previously approved drugs are
also referred to as listed drugs). Because the safety and efficacy
of listed drugs have already been established by the brand company
(sometimes referred to as the innovator), the FDA does not require
a demonstration of safety and efficacy of generic products.
However, a generic manufacturer is typically required to conduct
bioequivalence studies of its test product against the listed drug.
The bioequivalence studies for orally administered, systemically
available drug products assess the rate and extent to which the API
is absorbed into the bloodstream from the drug product and becomes
available at the site of action. Bioequivalence is established when
there is an absence of a significant difference in the rate and
extent for absorption of the generic product and the listed drug.
In addition to the bioequivalence data, an ANDA must contain patent
certifications and chemistry, manufacturing, labeling and stability
data.
The
third alternative is a special type of NDA, commonly referred to as
a Section 505(b)(2) NDA, which enables the applicant to rely,
in part, on the FDA's findings of safety and efficacy of an
existing product, or published literature, in support of its
application. Section 505(b)(2) NDAs often provide an alternate
path to FDA approval for new or improved formulations or new uses
of previously approved products. Section 505(b)(2) permits the
filing of an NDA where at least some of the information required
for approval comes from studies not conducted by or for the
applicant and for which the applicant has not obtained a right of
reference. The applicant may rely upon the FDA's findings with
respect to certain preclinical or clinical studies conducted for an
approved product. The FDA may also require companies to perform
additional studies or measurements to support the change from the
approved product. The FDA may then approve the new product
candidate for all or some of the label indications for which the
referenced product has been approved, as well as for any new
indication sought by the Section 505(b)(2)
applicant.
In
seeking approval for a drug through an NDA, including a 505(b)(2)
NDA, applicants are required to list with the FDA certain patents
of the applicant or that are held by third parties whose claims
cover the applicant's product. Upon approval of an NDA, each of the
patents listed in the application for the drug is then published in
the Orange Book. Any subsequent applicant who files an ANDA seeking
approval of a generic equivalent version of a drug listed in the
Orange Book or a 505(b)(2) NDA referencing a drug listed in the
Orange Book must make one of the following certifications to the
FDA concerning patents: (1) the patent information concerning
the reference listed drug product has not been submitted to the
FDA; (2) any such patent that was filed has expired;
(3) the date on which such patent will expire; or
(4) such patent is invalid or will not be infringed upon by
the manufacture, use or sale of the drug product for which the
application is submitted. This last certification is known as a
paragraph IV certification. A notice of the paragraph IV
certification must be provided to each owner of the patent that is
the subject of the certification and to the holder of the approved
NDA to which the ANDA or 505(b)(2) application refers. The
applicant may also elect to submit a "section viii" statement
certifying that its proposed label does not contain (or carves out)
any language regarding the patented method-of-use rather than
certify to a listed method-of-use patent.
If the
reference NDA holder or patent owners assert a patent challenge
directed to one of the Orange Book listed patents within
45 days of the receipt of the paragraph IV certification
notice, the FDA is prohibited from approving the application until
the earlier of 30 months from the receipt of the
paragraph IV certification expiration of the patent,
settlement of the lawsuit or a decision in the infringement case
that is favorable to the applicant. The ANDA or 505(b)(2)
application also will not be approved until any applicable
non-patent exclusivity listed in the Orange Book for the branded
reference drug has expired as described in further detail below.
Thus approval of a Section 505(b)(2) NDA or ANDA can be
stalled until all the listed patents claiming the referenced
product have expired, until any non-patent exclusivity, such as
exclusivity for obtaining approval of a new chemical entity, listed
in the Orange Book for the referenced product has expired, and, in
the case of a Paragraph IV certification and subsequent patent
infringement suit, until the earlier of 30 months, settlement
of the lawsuit or a decision in the infringement case that is
favorable to the ANDA or Section 505(b)(2)
applicant.
Post-Approval Requirements
Drugs
and biologics manufactured or distributed pursuant to FDA approvals
are subject to extensive and continuing regulation by the FDA,
including, among other things, requirements relating to
recordkeeping (including certain electronic record and signature
requirements), periodic reporting, product sampling and
distribution, advertising and promotion and reporting of certain
adverse experiences, deviations, and other problems with the
product. After approval, most changes to the approved product, such
as adding new indications or other labeling claims are subject to
prior FDA review and approval. There also are continuing, annual
user fee requirements for any marketed products and the
establishments at which such products are manufactured, as well as
new application fees for supplemental applications with clinical
data.
The FDA
strictly regulates labeling, advertising, promotion and other types
of information on products that are placed on the market. Products
may be promoted only for the approved indications and in accordance
with the provisions of the approved label. The FDA and other
agencies actively enforce the laws and regulations prohibiting the
promotion of off-label uses, and a company that is found to have
improperly promoted off-label uses may be subject to significant
liability. Further, manufacturers must continue to comply with cGMP
requirements, which are extensive and require considerable time,
resources and ongoing investment to ensure compliance. In addition,
changes to the manufacturing process generally require prior FDA
approval before being implemented and other types of changes to the
approved product, such as adding new indications and additional
labeling claims, are also subject to further FDA review and
approval.
Manufacturers
and certain other entities involved in the manufacturing and
distribution of approved products are required to register their
establishments with the FDA and certain state agencies, and are
subject to periodic unannounced inspections by the FDA and certain
state agencies for compliance with cGMP and other laws. The cGMP
requirements apply to all stages of the manufacturing process,
including the production, processing, sterilization, packaging,
labeling, storage and shipment of the product.
Changes
to the manufacturing process are strictly regulated and often
require prior FDA approval before being implemented. FDA
regulations also require investigation and correction of any
deviations from cGMP and impose reporting and documentation
requirements upon the sponsor and any third-party manufacturers
that the sponsor may decide to use. Accordingly, manufacturers must
continue to expend time, money, and effort in the area of
production and quality control to maintain cGMP
compliance.
The FDA
may impose a number of post-approval requirements as a condition of
approval of an application. For example, the FDA may require
post-marketing testing, including Phase 4 clinical trials, and
surveillance to further assess and monitor the product's safety and
effectiveness after commercialization.
The FDA
may withdraw a product approval if compliance with regulatory
requirements is not maintained or if problems occur after the
product reaches the market. Later discovery of previously unknown
problems with a product, including adverse events of unanticipated
severity or frequency, problems with manufacturing processes, or
failure to comply with regulatory requirements, may result in
restrictions on the product or even complete withdrawal of the
product from the market.
Potential
implications include required revisions to the approved labeling to
add new safety information; imposition of post-market studies or
clinical trials to assess new safety risks; or imposition of
distribution or other restrictions under a REMS program. Other
potential consequences include, among other things:
●
restrictions on the
marketing or manufacturing of the product, complete withdrawal of
the product from the market or product recalls;
●
warning letters or
holds on post-approval clinical trials;
●
refusal of the FDA
to approve pending NDAs/BLAs or supplements to approved NDAs/BLAs,
or suspension or revocation of product license
approvals;
●
product seizure or
detention, or refusal to permit the import or export of products;
or
●
injunctions or the
imposition of civil or criminal penalties.
In
addition, the distribution of prescription drugs and biologics is
subject to the Prescription Drug Marketing Act (PDMA), which
regulates the distribution of the products and product samples at
the federal level, and sets minimum standards for the registration
and regulation of distributors by the states. Both the PDMA and
state laws limit the distribution of prescription pharmaceutical
product samples and impose requirements to ensure accountability in
distribution.
From
time to time, legislation is drafted, introduced and passed in
Congress that could significantly change the statutory provisions
governing the approval, manufacturing and marketing of products
regulated by the FDA. In addition to new legislation, FDA
regulations, guidance, and policies are often revised or
reinterpreted by the agency in ways that may significantly affect
our business and our product candidates. It is impossible to
predict whether further legislative or FDA regulation or policy
changes will be enacted or implemented and what the impact of such
changes, if any, may be.
Pharmaceutical Coverage, Pricing and Reimbursement
In the
United States, sales of any products for which we may receive
regulatory approval for commercial sale will depend in part on the
availability of coverage and reimbursement from third-party payors.
Third-party payors include government authorities, managed care
providers, private health insurers and other organizations. The
process for determining whether a payor will provide coverage for a
drug may be separate from the process for setting the reimbursement
rate that the payor will pay for the product. Some of the
additional requirements and restrictions on coverage and
reimbursement levels imposed by third-party payors influence the
purchase of healthcare services and products. Third-party payors
may limit coverage to specific drugs on an approved list, or
formulary, which might not include all of the FDA-approved drugs
for a particular indication, or place drugs at certain formulary
levels that result in lower reimbursement levels and higher
cost-sharing obligation imposed on patients. Moreover, a payor's
decision to provide coverage for a drug product does not imply that
an adequate reimbursement rate will be approved. Adequate
third-party reimbursement may not be available to enable us to
maintain price levels sufficient to realize an appropriate return
on our investment in product development. Further, one payor's
determination to provide coverage does not assure that other payors
will also provide coverage and reimbursement for the product, and
the level of coverage and reimbursement may differ significantly
from payor to payor.
Third-party
payors are increasingly challenging the price and examining the
medical necessity and cost-effectiveness of medical products and
services, in addition to their safety and efficacy. In order to
obtain and maintain coverage and reimbursement for any product that
might be approved for sale, we may need to conduct expensive
pharmacoeconomic studies in order to demonstrate the medical
necessity and cost-effectiveness of any products, in addition to
the costs required to obtain regulatory approvals. Our product
candidates may not be considered medically necessary or
cost-effective. If third-party payors do not consider a product to
be cost-effective compared to other available therapies, they may
not cover the product after approval as a benefit under their plans
or, if they do, the level of payment may not be sufficient to allow
a company to sell its products at a profit.
The
U.S. government and state legislatures have shown significant
interest in implementing cost containment programs to limit the
growth of government-paid healthcare costs, including price
controls, restrictions on reimbursement and coverage and
requirements for substitution of generic products for branded
prescription drugs. Adoption of government controls and measures
and tightening of restrictive policies in jurisdictions with
existing controls and measures, could exclude or limit our drugs
and product candidates from coverage and limit payments for
pharmaceuticals.
In
addition, we expect that the increased emphasis on managed care and
cost containment measures in the United States by third-party
payors and government authorities to continue and will place
pressure on pharmaceutical pricing and coverage. Coverage policies
and third-party reimbursement rates may change at any time. Even if
favorable coverage and reimbursement status is attained for one or
more products for which we receive regulatory approval, less
favorable coverage policies and reimbursement rates may be
implemented in the future.
Employees
As of
December 31, 2017, we had five employees. Many of our activities
are outsourced to consultants who provide services to us on a
project basis. As business activities require and capital resources
permit, we will hire additional employees to fulfill our
company’s needs.
Form and Year of Organization
In 1965, the corporate predecessor of GT Biopharma, Diagnostic
Data, Inc. was incorporated in the State of California. Diagnostic
Data changed its incorporation to the State of Delaware in 1972;
and changed its name to DDI Pharmaceuticals, Inc. in 1985. In 1994,
DDI Pharmaceuticals merged with International BioClinical, Inc. and
Bioxytech S.A. and changed its name to OXIS International, Inc. On
July 17, 2017, we amended our Certificate of Incorporation for the
purpose of changing our name from Oxis International, Inc. to GT
Biopharma, Inc.
Agreement and Plan of Merger
On September 1, 2017, we entered into an Agreement and Plan of
Merger whereby we acquired 100% of the issued and outstanding
capital stock of GTP. GTP is a biotechnology company focused on
acquiring or discovering and patenting what it believes to be
close-to-market improved treatments for CNS disease (neurology and
pain) and shepherding the products through the FDA approval process
potentially to NDA. GTP products currently include a treatment for
neuropathic pain, the symptoms of myasthenia gravis, and motion
sickness. In exchange for the ownership of GTP, we issued a total
of 16,927,878 shares of our common stock to the three prior owners
of GTP, which represented 33% of our issued and outstanding capital
stock on a fully diluted basis at the time of closing.
Upon the consummation of the acquisition, Anthony J. Cataldo
resigned as our chief executive officer and was simultaneously
elected as executive chairman of the board of directors. Kathleen
Clarence-Smith, M.D., Ph.D., the founder of GTP, was then elected
as our chief executive officer and a member of the board of
directors.
As conditions to the acquisition of GTP, (i) we raised $4,540,000
upon the sale of debentures which were subsequently converted into
3,575,109 shares of restricted common stock and 208,224 shares of
Series J Preferred Stock to a total of nine persons or entities;
(ii) canceled debt in the amount $17,295,352 upon the issuance of
13,712,516 shares of common stock and 700,278 shares of Series J
Preferred Stock to a total of 26 persons or entities; (iii) issued
494,911 shares of common stock and 5,046 shares of Series J
Preferred Stock upon the cashless exercise of warrants to a total
of 22 persons or entities; and (iv) converted 25,000 shares of
Series H Preferred Stock and 1,666,667 Series I Preferred Stock
into 5,327,734 shares of common stock held by a total of three
persons or entities. All stock issuances were exempt from the
registration requirements of Section 5 of the Act of 1933, as
amended, or the Act, pursuant to Section 4(a)(2) of the Act because
the issuances did not involve any public offering.
Investing in our common stock involves a high degree of risk. You
should carefully consider the risks and uncertainties described
below in addition to the other information contained in this
prospectus before deciding whether to invest in shares of our
common stock. If any of the following risks actually occur, our
business, financial condition or operating results could be harmed.
In that case, the trading price of our common stock could decline
and you may lose part or all of your investment. In the opinion of
management, the risks discussed below represent the material risks
known to the company. Additional risks and uncertainties not
currently known to us or that we currently deem immaterial may also
impair our business, financial condition and operating results and
adversely affect the market price of our common stock.
Risks Related to Our Business
Our business is at an early stage of development and we may not
develop therapeutic products that can be
commercialized.
Our
business is at an early stage of development. We do not have
immune-oncology products in late stage clinical trials and have
only recently begun clinical trials for our CNS product candidates.
We are still in the early stages of identifying and conducting
research on potential therapeutic products. Our potential
therapeutic products will require significant research and
development and pre-clinical and clinical testing prior to
regulatory approval in the United States and other countries. We
may not be able to obtain regulatory approvals, enter clinical
trials for any of our product candidates, or commercialize any
products. Our product candidates may prove to have undesirable and
unintended side effects or other characteristics adversely
affecting their safety, efficacy or cost effectiveness that could
prevent or limit their use. Any product using any of our technology
may fail to provide the intended therapeutic benefits or
achieve therapeutic benefits equal to or better than the standard
of treatment at the time of testing or production.
We have a history of operating losses and we expect to continue to
incur losses for the foreseeable future and we may never generate
revenue or achieve profitability.
As of
December 31, 2017, we had an accumulated deficit of $267,896,000.
We have not generated any significant revenue to date and are not
profitable, and have incurred losses in each year since our
inception. We do not expect to generate any product sales or
royalty revenues for at least four years. We expect to incur
significant additional operating losses for the foreseeable future
as we expand research and development and clinical trial
efforts.
Our
ability to achieve long-term profitability is dependent upon
obtaining regulatory approvals for our products and successfully
commercializing our products alone or with third parties. However,
our operations may not be profitable even if any of our products
under development are successfully developed and produced and
thereafter commercialized. Even if we achieve profitability in the
future, we may not be able to sustain profitability in subsequent
periods.
Even if
we succeed in commercializing one or more of our product
candidates, we expect to continue to incur substantial research and
development and other expenditures to develop and market additional
product candidates. The size of our future net losses will depend,
in part, on the rate of future growth of our expenses and our
ability to generate revenue. Our prior losses and expected future
losses have had and will continue to have an adverse effect on our
stockholders’ equity and working capital.
We will need additional capital to conduct our operations and
develop our products, and our ability to obtain the necessary
funding is uncertain.
We have
used a significant amount of cash since inception to finance the
continued development and testing of our product candidates, and we
expect to need substantial additional capital resources in order to
develop our product candidates going forward and launch and
commercialize any product candidates for which we receive
regulatory approval.
We may
not be successful in generating and/or maintaining operating cash
flow, and the timing of our capital expenditures and other
expenditures may not result in cash sufficient to sustain our
operations through the next 12 months. If financing is not
sufficient and additional financing is not available or available
only on terms that are detrimental to our long-term survival, it
could have a material adverse effect on our ability to continue to
function. The timing and degree of any future capital requirements
will depend on many factors, including:
●
the accuracy of the
assumptions underlying our estimates for capital needs in 2018 and
beyond;
●
scientific and
clinical progress in our research and development
programs;
●
the magnitude and
scope of our research and development programs and our ability to
establish, enforce and maintain strategic arrangements for
research, development, clinical testing, manufacturing and
marketing;
●
our progress with
pre-clinical development and clinical trials;
●
the time and costs
involved in obtaining regulatory approvals;
●
the costs involved
in preparing, filing, prosecuting, maintaining, defending and
enforcing patent claims; and
●
the number and type
of product candidates that we pursue.
Additional
financing through strategic collaborations, public or private
equity or debt financings or other financing sources may not be
available on acceptable terms, or at all. Additional equity
financing could result in significant dilution to our stockholders,
and any debt financings will likely involve covenants restricting
our business activities. Additional financing may not be available
on acceptable terms, or at all. Further, if we obtain additional
funds through arrangements with collaborative partners, these
arrangements may require us to relinquish rights to some of our
technologies, product candidates or products that we would
otherwise seek to develop and commercialize on our
own.
If
sufficient capital is not available, we may be required to delay,
reduce the scope of or eliminate one or more of our research or
product development initiatives, any of which could have a material
adverse effect on our financial condition or business
prospects.
We have identified material weaknesses in our internal control over
financial reporting have not remedied these weaknesses. If we fail
to maintain an effective system of internal control over financial
reporting, we may not be able to accurately report our financial
results or prevent fraud. As a result, stockholders could lose
confidence in our financial and other public reporting, which would
harm our business and the trading price of our common
stock.
Effective
internal control over financial reporting is necessary for us to
provide reliable financial reports and, together with adequate
disclosure controls and procedures, are designed to prevent fraud.
Any failure to implement required new or improved controls, or
difficulties encountered in their implementation, could cause us to
fail to meet our reporting obligations. Ineffective internal
control could also cause investors to lose confidence in our
reported financial information, which could have a negative effect
on the trading price of our common stock.
We have identified material weaknesses in our internal control over
financial reporting as a company. As defined in Regulation 12b-2
under the Securities Exchange Act of 1934, or the Exchange Act, a
“material weakness” is a deficiency, or combination of
deficiencies, in internal control over financial reporting, such
that there is a reasonable possibility that a material misstatement
of our annual or interim consolidated financial statements will not
be prevented, or detected on a timely basis. Specifically, we
determined that we had the following material weaknesses in our
internal control over financial reporting: (i) inadequate
segregation of duties; and (ii) insufficient written policies and
procedures for accounting and financial reporting with respect to
the requirements and application of both generally accepted
accounting principles in the United States of America, or GAAP, and
the U.S. Securities and Exchange Commission, or the SEC,
guidelines.
As of the date of this report, we have not remediated these
material weaknesses. We are continuing to adopt and implement
written policies and procedures for accounting and financial
reporting. We plan to hire additional qualified personnel to
address inadequate segregation of duties, although the timing of
such hires is largely dependent on our securing additional
financing to cover such costs. The implementation of these
initiatives may not fully address any material weakness or other
deficiencies that we may have in our internal control over
financial reporting.
Even if we develop effective internal control over financial
reporting, such controls may become inadequate due to changes in
conditions or the degree of compliance with such policies or
procedures may deteriorate, which could result in the discovery of
additional material weaknesses and deficiencies. In any event, the
process of determining whether our existing internal control over
financial reporting is compliant with Section 404 of the
Sarbanes-Oxley Act, or Section 404, and sufficiently effective
requires the investment of substantial time and resources,
including by certain members of our senior management. As a result,
this process may divert internal resources and take a significant
amount of time and effort to complete. In addition, we cannot
predict the outcome of this process and whether we will need to
implement remedial actions in order to establish effective controls
over financial reporting. The determination of whether or not our
internal controls are sufficient and any remedial actions required
could result in us incurring additional costs that we did not
anticipate, including the hiring of outside consultants. We may
also fail to timely complete our evaluation, testing and any
remediation required to comply with Section 404.
We are required, pursuant to Section 404, to furnish a report by
management on, among other things, the effectiveness of our
internal control over financial reporting. However, for as long as
we are a “smaller reporting company,” our independent
registered public accounting firm will not be required to attest to
the effectiveness of our internal control over financial reporting
pursuant to Section 404. While we could be a smaller reporting
company for an indefinite amount of time, and thus relieved of the
above-mentioned attestation requirement, an independent assessment
of the effectiveness of our internal control over financial
reporting could detect problems that our management's assessment
might not. Such undetected material weaknesses in our internal
control over financial reporting could lead to financial statement
restatements and require us to incur the expense of
remediation.
Our intellectual property may be compromised.
Part of
our value going forward depends on the intellectual property rights
that we have been and are acquiring. There may have been many
persons involved in the development of our intellectual property,
and we may not be successful in obtaining the necessary rights from
all of them. It is possible that in the future, third parties may
challenge our intellectual property rights. We may not be
successful in protecting our intellectual property rights. In
either event, we may lose the value of our intellectual property,
and if so, our business prospects may suffer.
If our efforts to protect the proprietary nature of the
intellectual property related to our technologies are not adequate,
we may not be able to compete effectively in our market and our
business would be harmed.
We rely
upon a combination of patents, trade secret protection and
confidentiality agreements to protect the intellectual property
related to our technologies. Any disclosure to or misappropriation
by third parties of our trade secret or other confidential
information could enable competitors to quickly duplicate or
surpass our technological achievements, thus eroding any
competitive advantage we may derive from this
information.
The
strength of patents in the biotechnology and pharmaceutical field
involves complex legal and scientific questions and can be
uncertain. The patent applications we own or license may fail to
result in issued patents in the United States or in foreign
countries. Third parties may challenge the validity, enforceability
or scope of any issued patents we own or license or any
applications that may issue as patents in the future, which may
result in those patents being narrowed, invalidated or held
unenforceable. Even if they are unchallenged, our patents and
patent applications may not adequately protect our intellectual
property or prevent others from developing similar products that do
not fall within the scope of our patents. If the breadth or
strength of protection provided by the patents we hold or pursue is
threatened, our ability to commercialize any product candidates
with technology protected by those patents could be threatened.
Further, if we encounter delays in our clinical trials, the period
of time during which we would have patent protection for any
covered product candidates that obtain regulatory approval would be
reduced. Since patent applications in the United States and most
other countries are confidential for a period of time after filing,
we cannot be certain at the time of filing that we are the first to
file any patent application related to our product
candidates.
In
addition to the protection afforded by patents, we seek to rely on
trade secret protection and confidentiality agreements to protect
proprietary know-how that is not patentable, processes for which
patents are difficult to enforce and any other elements of our
discovery platform and drug development processes that involve
proprietary know-how, information or technology that is not covered
by patents or not amenable to patent protection. Although we
require all of our employees and certain consultants and advisors
to assign inventions to us, and all of our employees, consultants,
advisors and any third parties who have access to our proprietary
know-how, information or technology to enter into confidentiality
agreements, our trade secrets and other proprietary information may
be disclosed or competitors may otherwise gain access to such
information or independently develop substantially equivalent
information. Further, the laws of some foreign countries do not
protect proprietary rights to the same extent or in the same manner
as the laws of the United States. As a result, we may encounter
significant difficulty in protecting and defending our intellectual
property both in the United States and abroad. If we are unable to
prevent material disclosure of the trade secret intellectual
property related to our technologies to third parties, we may not
be able to establish or maintain the competitive advantage that we
believe is provided by such intellectual property, which could
materially adversely affect our market position and business and
operational results.
Claims that we infringe the intellectual property rights of others
may prevent or delay our drug discovery and development
efforts.
Our
research, development and commercialization activities, as well as
any product candidates or products resulting from those activities,
may infringe or be accused of infringing a patent or other form of
intellectual property under which we do not hold a license or other
rights. Third parties may assert that we are employing their
proprietary technology without authorization. There may be
third-party patents of which we are currently unaware of, with
claims that cover the use or manufacture of our product candidates
or the practice of our related methods. Because patent applications
can take many years to issue, there may be currently pending patent
applications that may later result in issued patents that our
product candidates may infringe. In addition, third parties may
obtain patents in the future and claim that use of our technologies
infringes one or more claims of these patents. If our activities or
product candidates infringe the patents or other intellectual
property rights of third parties, the holders of such intellectual
property rights may be able to block our ability to commercialize
such product candidates or practice our methods unless we obtain a
license under the intellectual property rights or until any
applicable patents expire or are determined to be invalid or
unenforceable.
Defense
of any intellectual property infringement claims against us,
regardless of their merit, would involve substantial litigation
expense and would be a significant diversion of employee resources
from our business. In the event of a successful claim of
infringement against us, we may have to pay substantial damages,
obtain one or more licenses from third parties, limit our business
to avoid the infringing activities, pay royalties and/or redesign
our infringing product candidates or methods, any or all of which
may be impossible or require substantial time and monetary
expenditure. Further, if we were to seek a license from the third
party holder of any applicable intellectual property rights, we may
not be able to obtain the applicable license rights when needed or
on commercially reasonable terms, or at all. The occurrence of any
of the above events could prevent us from continuing to develop and
commercialize one or more of our product candidates and our
business could materially suffer.
We may desire, or be forced, to seek additional licenses to use
intellectual property owned by third parties, and such licenses may
not be available on commercially reasonable terms or at
all.
A third
party may hold intellectual property, including patent rights, that
are important or necessary to the development of our product
candidates, in which case we would need to obtain a license from
that third party or develop a different formulation of the product
that does not infringe upon the applicable intellectual property,
which may not be possible. Additionally, we may identify product
candidates that we believe are promising and whose development and
other intellectual property rights are held by third parties. In
such a case, we may desire to seek a license to pursue the
development of those product candidates. Any license that we may
desire to obtain or that we may be forced to pursue may not be
available when needed on commercially reasonable terms or at all.
Any inability to secure a license that we need or desire could have
a material adverse effect on our business, financial condition and
prospects.
The patent protection covering some of our product candidates may
be dependent on third parties, who may not effectively maintain
that protection.
While
we expect that we will generally seek to gain the right to fully
prosecute any patents covering product candidates we may in-license
from third-party owners, there may be instances when platform
technology patents that cover our product candidates remain
controlled by our licensors. If any of our current or future
licensing partners that retain the right to prosecute patents
covering the product candidates we license from them fail to
appropriately maintain that patent protection, we may not be able
to prevent competitors from developing and selling competing
products or practicing competing methods and our ability to
generate revenue from any commercialization of the affected product
candidates may suffer.
We may be involved in lawsuits to protect or enforce our patents or
the patents of our licensors, which could be expensive,
time-consuming and unsuccessful.
Competitors
may infringe our patents or the patents of our current or potential
licensors. To attempt to stop infringement or unauthorized use, we
may need to enforce one or more of our patents, which can be
expensive and time-consuming and distract management. If we pursue
any litigation, a court may decide that a patent of ours or our
licensor’s is not valid or is unenforceable, or may refuse to
stop the other party from using the relevant technology on the
grounds that our patents do not cover the technology in question.
Further, the legal systems of certain countries, particularly
certain developing countries, do not favor the enforcement of
patents, which could reduce the likelihood of success of any
infringement proceeding we pursue in any such jurisdiction. An
adverse result in any infringement litigation or defense
proceedings could put one or more of our patents at risk of being
invalidated, held unenforceable, or interpreted narrowly and could
put our patent applications at risk of not issuing, which could
limit our ability to exclude competitors from directly competing
with us in the applicable jurisdictions.
Interference
proceedings provoked by third parties or brought by the U.S. PTO
may be necessary to determine the priority of inventions with
respect to our patents or patent applications or those of our
licensors. An unfavorable outcome could require us to cease using
the related technology or to attempt to license rights to use it
from the prevailing party. Our business could be harmed if the
prevailing party does not offer us a license on commercially
reasonable terms, or at all. Litigation or interference proceedings
may fail and, even if successful, may result in substantial costs
and distract our management and other employees.
If we are unsuccessful in obtaining or maintaining patent
protection for intellectual property in development, our business
and competitive position would be harmed.
We are
seeking patent protection for some of our technology and product
candidates. Patent prosecution is a challenging process and is not
assured of success. If we are unable to secure patent protection
for our technology and product candidates, our business may be
adversely impacted.
In
addition, issued patents and pending international applications
require regular maintenance. Failure to maintain our portfolio may
result in loss of rights that may adversely impact our intellectual
property rights, for example by rendering issued patents
unenforceable or by prematurely terminating pending international
applications.
If we are unable to protect the confidentiality of our trade
secrets, our business and competitive position would be
harmed.
In
addition to seeking patents for some of our technology and product
candidates, we also rely on trade secrets, including unpatented
know-how, technology and other proprietary information, to maintain
our competitive position. We currently, and expect in the future to
continue to, seek to protect these trade secrets, in part, by
entering into confidentiality agreements with parties who have
access to them, such as our employees, collaborators, contract
manufacturers, consultants, advisors and other third parties. We
also enter into confidentiality and invention or patent assignment
agreements with our employees and consultants. Despite these
efforts, any of these parties may breach the agreements and
disclose our proprietary information, including our trade secrets,
and we may not be able to obtain adequate remedies for any such
disclosure. Enforcing a claim that a party illegally disclosed or
misappropriated a trade secret is difficult, expensive and
time-consuming, and the outcome is unpredictable. In addition, some
courts inside and outside the United States are less willing or
unwilling to protect trade secrets. If any of our trade secrets
were to be lawfully obtained or independently developed by a
competitor, we would have no right to prevent them, or those to
whom they disclose the trade secrets, from using that technology or
information to compete with us. If any of our trade secrets were to
be disclosed to or independently developed by a competitor, our
competitive position would be harmed.
If we fail to meet our obligations under our license agreements, we
may lose our rights to key technologies on which our business
depends.
Our
business depends in part on licenses from third parties. These
third-party license agreements impose obligations on us, such as
payment obligations and obligations to diligently pursue
development of commercial products under the licensed patents. If a
licensor believes that we have failed to meet our obligations under
a license agreement, the licensor could seek to limit or terminate
our license rights, which could lead to costly and time-consuming
litigation and, potentially, a loss of the licensed rights. During
the period of any such litigation, our ability to carry out the
development and commercialization of potential products could be
significantly and negatively affected. If our license rights were
restricted or ultimately lost, our ability to continue our business
based on the affected technology platform could be severely
adversely affected.
We will have to hire additional executive officers and employees to
operate our business. If we are unable to hire qualified personnel,
we may not be able to implement our business strategy.
We
currently have only five fulltime employees. The loss of the
services of any of our key product or business development
employees could delay our product development programs and our
research and development efforts. We do not maintain key person
life insurance on any of our officers, employees or consultants. In
order to develop our business in accordance with our business
strategy, we will have to hire additional qualified personnel,
including in the areas of manufacturing, clinical trials
management, regulatory affairs, and business development. We will
need to raise sufficient funds to hire the necessary employees and
have commenced our search for additional key
employees.
Moreover,
there is intense competition for a limited number of qualified
personnel among biopharmaceutical, biotechnology, pharmaceutical
and other businesses. Many of the other pharmaceutical companies
against which we compete for qualified personnel have greater
financial and other resources, different risk profiles, longer
histories in the industry and greater ability to provide valuable
cash or stock incentives to potential recruits than we do. They
also may provide more diverse opportunities and better chances for
career advancement. Some of these characteristics may be more
appealing to high quality candidates than what we are able to offer
as an early-stage company. If we are unable to continue to attract
and retain high quality personnel, the rate and success at which we
can develop and commercialize product candidates will be
limited.
We depend on key personnel for our continued operations and future
success, and a loss of certain key personnel could significantly
hinder our ability to move forward with our business
plan.
Because
of the specialized nature of our business, we are highly dependent
on our ability to identify, hire, train and retain highly qualified
scientific and technical personnel for the research and development
activities we conduct or sponsor. The loss of one or more key
executive officers, or scientific officers, would be significantly
detrimental to us. In addition, recruiting and retaining qualified
scientific personnel to perform research and development work is
critical to our success. Our anticipated growth and expansion into
areas and activities requiring additional expertise, such as
clinical testing, regulatory compliance, manufacturing and
marketing, will require the addition of new management personnel
and the development of additional expertise by existing management
personnel. There is intense competition for qualified personnel in
the areas of our present and planned activities. Accordingly, we
may not be able to continue to attract and retain the qualified
personnel, which would adversely affect the development of our
business.
We may be subject to claims by third parties asserting that our
employees or we have misappropriated their intellectual property,
or claiming ownership of what we regard as our own intellectual
property.
Many of
our employees were previously employed at universities or other
biotechnology or pharmaceutical companies, including our
competitors or potential competitors. Although we try to ensure
that our employees do not use the proprietary information or
know-how of others in their work for us, with contractual
provisions and other procedures, we may be subject to claims that
these employees or we have used or disclosed intellectual property,
including trade secrets or other proprietary information, of any
such employee’s former employers. Litigation may be necessary
to defend against any such claims.
In
addition, while it is our policy to require our employees and
contractors who may be involved in the development of intellectual
property to execute agreements assigning such intellectual property
to us, we may be unsuccessful in executing such an agreement with
each party who in fact contributes to the development of
intellectual property that we regard as our own. Further, the terms
of such assignment agreements may be breached and we may not be
able to successfully enforce their terms, which may force us to
bring claims against third parties, or defend claims they may bring
against us, to determine the ownership of intellectual property
rights we may regard and treat as our own.
Our employees may engage in misconduct or other improper
activities, including noncompliance with regulatory standards and
requirements, which could cause our business to
suffer.
We are
exposed to the risk of employee fraud or other misconduct.
Misconduct by employees could include intentional failures to
comply with regulations of governmental authorities, such as the
FDA or the European Medicines Agency, or EMA, to provide accurate
information to the FDA or EMA, to comply with manufacturing
standards we have established, to comply with federal, state and
international healthcare fraud and abuse laws and regulations as
they may become applicable to our operations, to report financial
information or data accurately or to disclose unauthorized
activities to us. Employee misconduct could also involve the
improper use of information obtained in the course of clinical
trials, which could result in regulatory sanctions and serious harm
to our reputation. It is not always possible to identify and deter
employee misconduct, and the precautions we currently take and the
procedures we may establish in the future as our operations and
employee base expand to detect and prevent this type of activity
may not be effective in controlling unknown or unmanaged risks or
losses or in protecting us from governmental investigations or
other actions or lawsuits stemming from a failure by our employees
to comply with such laws or regulations. If any such actions are
instituted against us, and we are not successful in defending
ourselves or asserting our rights, those actions could have a
significant impact on our business and results of operations,
including the imposition of significant fines or other
sanctions.
Our reliance on the activities of our non-employee consultants,
research institutions and scientific contractors, whose activities
are not wholly within our control, may lead to delays in
development of our proposed products.
We rely
extensively upon and have relationships with scientific consultants
at academic and other institutions, some of whom conduct research
at our request, and other consultants with expertise in clinical
development strategy or other matters. These consultants are not
our employees and may have commitments to, or consulting or
advisory contracts with, other entities that may limit their
availability to us. We have limited control over the activities of
these consultants and, except as otherwise required by our
collaboration and consulting agreements to the extent they exist,
can expect only limited amounts of their time to be dedicated to
our activities. These research facilities may have commitments to
other commercial and non-commercial entities. We have limited
control over the operations of these laboratories and can expect
only limited amounts of time to be dedicated to our research
goals.
It may take longer to complete our clinical trials than we project,
or we may not be able to complete them at all.
For
budgeting and planning purposes, we have projected the date for the
commencement, continuation and completion of our various clinical
trials. However, a number of factors, including scheduling
conflicts with participating clinicians and clinical institutions,
and difficulties in identifying and enrolling patients who meet
trial eligibility criteria, may cause significant delays. We may
not commence or complete clinical trials involving any of our
products as projected or may not conduct them
successfully.
We
expect to rely on medical institutions, academic institutions or
clinical research organizations to conduct, supervise or monitor
some or all aspects of clinical trials involving our products. We
will have less control over the timing and other aspects of these
clinical trials than if we conducted them entirely on our own. If
we fail to commence or complete, or experience delays in, any of
our planned clinical trials, our stock price and our ability to
conduct our business as currently planned could be
harmed.
Clinical drug development is costly, time-consuming and uncertain,
and we may suffer setbacks in our clinical development program that
could harm our business.
Clinical
drug development for our product candidates is costly,
time-consuming and uncertain. Our product candidates are in various
stages of development and while we expect that clinical trials for
these product candidates will continue for several years, such
trials may take significantly longer than expected to complete. In
addition, we, the FDA, an institutional review board, or IRB, or
other regulatory authorities, including state and local agencies
and counterpart agencies in foreign countries, may suspend, delay,
require modifications to or terminate our clinical trials at any
time, for various reasons, including:
●
discovery of safety
or tolerability concerns, such as serious or unexpected toxicities
or side effects or exposure to otherwise unacceptable health risks,
with respect to study participants;
●
lack of
effectiveness of any product candidate during clinical trials or
the failure of our product candidates to meet specified
endpoints;
●
delays in subject
recruitment and enrollment in clinical trials or inability to
enroll a sufficient number of patients in clinical trials to ensure
adequate statistical ability to detect statistically significant
treatment effects;
●
difficulty in
retaining subjects and volunteers in clinical trials;
●
difficulty in
obtaining IRB approval for studies to be conducted at each clinical
trial site;
●
delays in
manufacturing or obtaining, or inability to manufacture or obtain,
sufficient quantities of materials for use in clinical
trials;
●
inadequacy of or
changes in our manufacturing process or the product formulation or
method of delivery;
●
delays or failure
in reaching agreement on acceptable terms in clinical trial
contracts or protocols with prospective contract research
organizations, or CROs, clinical trial sites and other third-party
contractors;
●
inability to add a
sufficient number of clinical trial sites;
●
uncertainty
regarding proper formulation and dosing;
●
failure by us, our
employees, our CROs or their employees or other third-party
contractors to comply with contractual and applicable regulatory
requirements or to perform their services in a timely or acceptable
manner;
●
scheduling
conflicts with participating clinicians and clinical
institutions;
●
failure to design
appropriate clinical trial protocols;
●
inability or
unwillingness of medical investigators to follow our clinical
protocols;
●
difficulty in
maintaining contact with subjects during or after treatment, which
may result in incomplete data; or
●
changes in
applicable laws, regulations and regulatory policies.
If we experience delays or difficulties in the enrollment of
patients in clinical trials, those clinical trials could take
longer than expected to complete and our receipt of necessary
regulatory approvals could be delayed or prevented.
We may
not be able to initiate or continue clinical trials for our product
candidates if we are unable to locate and enroll a sufficient
number of eligible patients to participate in these trials as
required by U.S. Food and Drug Administration, or the FDA, or
similar regulatory authorities outside the United States. In
particular, because we are focused on patients with molecularly
defined cancers, our pool of suitable patients may be smaller and
more selective and our ability to enroll a sufficient number of
suitable patients may be limited or take longer than anticipated.
In addition, some of our competitors have ongoing clinical trials
for product candidates that treat the same indications as our
product candidates, and patients who would otherwise be eligible
for our clinical trials may instead enroll in clinical trials of
our competitors’ product candidates.
Patient
enrollment for any of our clinical trials may also be affected by
other factors, including without limitation:
●
the severity of the
disease under investigation;
●
the frequency of
the molecular alteration we are seeking to target in the applicable
trial;
●
the eligibility
criteria for the study in question;
●
the perceived risks
and benefits of the product candidate under study;
●
the extent of the
efforts to facilitate timely enrollment in clinical
trials;
●
the patient
referral practices of physicians;
●
the ability to
monitor patients adequately during and after treatment;
and
●
the proximity and
availability of clinical trial sites for prospective
patients.
Our
inability to enroll a sufficient number of patients for our
clinical trials would result in significant delays and could
require us to abandon one or more clinical trials altogether.
Enrollment delays in our clinical trials may result in increased
development costs for our product candidates, and we may not have
or be able to obtain sufficient cash to fund such increased costs
when needed, which could result in the further delay or termination
of the trial.
Consistent
with our general product development strategy, we intend to design
future trials for our product candidates to include some patients
with the applicable clinical characteristics, stage of therapy,
molecular alterations, biomarkers, and/or cell surface antigens
that determine therapeutic options, or are indicators of the
disease, with a view to assessing possible early evidence of
potential therapeutic effect. If we are unable to locate and
include such patients in those trials, then our ability to make
those early assessments and to seek participation in FDA expedited
review and approval programs, including breakthrough therapy and
fast track designation, or otherwise to seek to accelerate clinical
development and regulatory timelines, could be
compromised.
We have limited clinical testing and regulatory capabilities, and
human clinical trials are subject to extensive regulatory
requirements, very expensive, time-consuming and difficult to
design and implement. Our products may fail to achieve necessary
safety and efficacy endpoints during clinical trials, which may
limit our ability to generate revenues from therapeutic
products.
We
cannot assure you that we will be able to invest or develop
resources for clinical trials successfully or as expediently as
necessary. In particular, human clinical trials can be very
expensive and difficult to design and implement, in part because
they are subject to rigorous regulatory requirements. The clinical
trial process is time consuming. We estimate that clinical trials
of our product candidates will take at least several years to
complete. Furthermore, failure can occur at any stage of the
trials, and we could encounter problems that cause us to abandon or
repeat clinical trials. The commencement and completion of clinical
trials may be affected by several factors, including:
●
unforeseen safety
issues;
●
determination of
dosing issues;
●
inability to
demonstrate effectiveness during clinical trials;
●
slower than
expected rates of patient recruitment;
●
inability to
monitor patients adequately during or after treatment;
and
●
inability or
unwillingness of medical investigators to follow our clinical
protocols.
In
addition, we or the FDA, may suspend our clinical trials at any
time if it appears that we are exposing participants to
unacceptable health risks or if the FDA finds deficiencies in our
investigational new drug application, or IND, submissions or the
conduct of these trials.
We are subject to extensive regulation, which can be costly and
time consuming and can subject us to unanticipated delays. even if
we obtain regulatory approval for some of our products, those
products may still face regulatory difficulties.
All of
our potential products, processing and manufacturing activities,
are subject to comprehensive regulation by the FDA in the United
States and by comparable authorities in other countries. The
process of obtaining FDA and other required regulatory approvals,
including foreign approvals, is expensive and often takes many
years and can vary substantially based upon the type, complexity
and novelty of the products involved. In addition, regulatory
agencies may lack experience with our technologies and products,
which may lengthen the regulatory review process, increase our
development costs and delay or prevent their
commercialization.
If we
violate regulatory requirements at any stage, whether before or
after we obtain marketing approval, the FDA may take enforcement
action(s) against us, which could include issuing a warning or
untitled letter, placing a clinical hold on an ongoing clinical
trial, product seizure, enjoining our operations, refusal to
consider our applications for pre-market approval, refusal of an
investigational new drug application, fines, or even civil or
criminal liability, any of which could materially harm our
reputation and financial results. Additionally, we may not be able
to obtain the labeling claims necessary or desirable for the
promotion of our products. We may also be required to undertake
postmarketing trials to provide additional evidence of safety
and effectiveness. In addition, if we or others identify side
effects after any of our adoptive therapies are on the market, or
if manufacturing problems occur, regulators may withdraw their
approval and reformulations, additional clinical trials, changes in
labeling of our products, and additional marketing applications may
be required.
Any of
the following factors, among others, could cause regulatory
approval for our product candidates to be delayed, limited or
denied:
●
the product
candidates require significant clinical testing to demonstrate
safety and effectiveness before applications for marketing approval
can be filed with the FDA and other regulatory
authorities;
●
data obtained from
pre-clinical and nonclinical animal testing and clinical trials can
be interpreted in different ways, and regulatory authorities may
not agree with our respective interpretations or may require us to
conduct additional testing;
●
negative or
inconclusive results or the occurrence of serious or unexpected
adverse events during a clinical trial could cause us to delay or
terminate development efforts for a product candidate;
and/or
●
FDA and other
regulatory authorities may require expansion of the size and scope
of the clinical trials.
Any
difficulties or failures that we encounter in securing regulatory
approval for our product candidates would likely have a substantial
adverse impact on our ability to generate product sales, and could
make any search for a collaborative partner more
difficult.
Obtaining regulatory approval even after clinical trials that are
believed to be successful is an uncertain process.
Even if
we complete our planned clinical trials and believe the results
were successful, obtaining regulatory approval is a lengthy,
expensive and uncertain process, and the FDA or other regulatory
agencies may delay, limit or deny approval of any of our
applications for pre-market approval for many reasons,
including:
●
we may not be able
to demonstrate to the FDA’s satisfaction that our product
candidates are safe and effective for any indication;
●
the results of
clinical trials may not meet the level of statistical significance
or clinical significance required by the FDA for
approval;
●
the FDA may
disagree with the number, design, size, conduct or implementation
of our clinical trials;
●
the FDA may not
find the data from pre-clinical studies and clinical trials
sufficient to demonstrate that the clinical and other benefits of
our product candidates outweigh their safety risks;
●
the FDA may
disagree with our interpretation of data from pre-clinical studies
or clinical trials, or may not accept data generated at our
clinical trial sites;
●
the data collected
from pre-clinical studies and clinical trials of our product
candidates may not be sufficient to support the submission of
applications for regulatory approval;
●
the FDA may have
difficulties scheduling an advisory committee meeting in a timely
manner, or the advisory committee may recommend against approval of
our application or may recommend that the FDA require, as a
condition of approval, additional pre-clinical studies or clinical
trials, limitations on approved labeling, or distribution and use
restrictions;
●
the FDA may require
development of a risk evaluation and mitigation strategy as a
condition of approval;
●
the FDA may
identify deficiencies in the manufacturing processes or facilities
of third-party manufacturers with which we enter into agreements
for clinical and commercial supplies;
●
the FDA may change
their approval policies or adopt new regulations that adversely
affect our applications for pre-market approval; and
●
the FDA may require
simultaneous approval for both adults and for children and
adolescents delaying needed approvals, or we may have successful
clinical trial results for adults but not children and adolescents,
or vice versa.
Before
we can submit an application for regulatory approval in the United
States, we must conduct a pivotal, Phase 3 trial. We will also need
to agree on a protocol with the FDA for a clinical trial before
commencing the trial. Phase 3 clinical trials frequently produce
unsatisfactory results even though prior clinical trials were
successful. Therefore, even if the results of our Phase 2 trials
are successful, the results of the additional trials that we
conduct may or may not be successful. Further, our product
candidates may not be approved even if they achieve their primary
endpoints in Phase 3 clinical trials. The FDA or other foreign
regulatory authorities may disagree with our trial design and our
interpretation of data from preclinical studies and clinical
trials. Any of these regulatory authorities may change requirements
for the approval of a product candidate even after reviewing and
providing comments or advice on a protocol for a clinical trial.
The FDA or other regulatory agencies may require that we conduct
additional clinical, nonclinical, manufacturing validation or drug
product quality studies and submit those data before considering or
reconsidering the application. Depending on the extent of these or
any other studies, approval of any applications that we submit may
be delayed by several years, or may require us to expend more
resources than we have available. It is also possible that
additional studies, if performed and completed, may not be
considered sufficient by the FDA or other regulatory
agencies.
In
addition, the FDA or other regulatory agencies may also approve a
product candidate for fewer or more limited indications than we
request, may impose significant limitations related to use
restrictions for certain age groups, warnings, precautions or
contraindications or may grant approval contingent on the
performance of costly post-marketing clinical trials or risk
mitigation requirements.
We intend to pursue Section 505(b)(2) regulatory approval
filings with the FDA for at least three of our product candidates.
If the FDA concludes that certain of our product candidates fail to
satisfy the requirements under Section 505(b)(2), or if the
requirements for such product candidates under
Section 505(b)(2) are not as we expect, the approval
pathway for such product candidates may take significantly longer,
cost substantially more and entail greater complications and risks
than anticipated and, in either case, may not be successful. In
addition, if under certain circumstances, exclusivity of
competitors would delay approval of our product candidates, then we
may pursue approval through the Section 505(b)(1) regulatory
pathway, which may require us to conduct additional preclinical or
clinical trials or obtain a right to reference the preclinical or
clinical data of others.
We are
currently developing three product candidates, GTP-004, GTP-011 and
PainBrake for which we intend to seek FDA approval through the
Section 505(b)(2) regulatory pathway, and may decide to
seek FDA approval for other products through the
Section 505(b)(2) regulatory pathway in the future. A
Section 505(b)(2) NDA is a special type of NDA that
enables the applicant to rely, in part, on the FDA’s findings
of safety and efficacy of an existing previously approved product,
or published literature, in support of its application.
Section 505(b)(2) NDAs often provide an alternate path to
FDA approval for new or improved formulations or new uses of
previously approved products. Such filings involve significant
filing costs, including filing fees.
Reliance
on existing safety findings could expedite the development program
for our product candidates by decreasing the amount of preclinical
or clinical data that we would need to generate in order to obtain
FDA approval. If the FDA does not allow us to pursue the
Section 505(b)(2) regulatory pathway as anticipated, or
if the Section 505(b)(2) regulatory pathway fails to
significantly decrease the amount of testing we must conduct, we
may need to conduct additional preclinical or clinical trials,
provide additional data and information and meet additional
standards to obtain regulatory approval. In such case, the time and
financial resources required to obtain FDA approval for product
candidates for which we seek approval through the
Section 505(b)(2) pathway in the future, and complications and
risks associated with these product candidates, likely would
increase substantially. Moreover, our inability to pursue the
Section 505(b)(2) regulatory pathway could prevent us
from introducing our product candidates into the market prior to
our competitors, which could harm our competitive position and
prospects. Even if the FDA allows us to pursue approval through the
Section 505(b)(2), we cannot guarantee that it would
ultimately lead to faster product development, and our product
candidates may not receive the requisite approvals for
commercialization.
Furthermore,
Section 505(b)(2) NDAs are subject to special
requirements designed to protect the patent rights of sponsors of
previously approved drugs referenced in a
Section 505(b)(2) NDA, and pursuing the Section 505(b)(2)
pathway could lead to patent litigation and other significant
delays if a current patent holder challenges our application for
pre-market approval. In addition, a manufacturer of an approved
referenced product to file a citizen petition with the FDA
seeking to delay approval of, or impose additional approval
requirements for, pending competing products. If successful, such
petitions can significantly delay, or even prevent, the approval of
the new product. However, even if the FDA ultimately denies such a
petition, the FDA may substantially delay approval while it
considers and responds to the petition.
Furthermore,
award of three-year exclusivity by the FDA to a competitor with a
Section 505(b)(2) NDA could delay approval of a product candidate
of ours submitted pursuant to Section 505(b)(2) of the Food, Drug,
and Cosmetic Act if the FDA were to determine that the products
have overlapping conditions of approval, even if our Section
505(b)(2) NDA does not rely on the competing Section 505(b)(2) NDA.
Alternatively, we may pursue approval through the Section 505(b)(1)
regulatory pathway, which may require us to conduct additional
preclinical or clinical trials or obtain a right to reference the
preclinical or clinical data of others. These alternatives may
increase the time and/or financial resources required to obtain
approval.
We will continue to be subject to extensive FDA regulation
following any product approvals, and if we fail to comply with
these regulations, we may suffer a significant setback in our
business.
Even if
we are successful in obtaining regulatory approval of our product
candidates, we will continue to be subject to the requirements of
and review by, the FDA and comparable regulatory authorities in the
areas of manufacturing processes, post-approval clinical data,
adverse event reporting, labeling, advertising and promotional
activities, among other things. In addition, any marketing approval
we receive may be limited in terms of the approved product
indication or require costly post-marketing testing and
surveillance. Discovery after approval of previously unknown
problems with a product, manufacturer or manufacturing process, or
a failure to comply with regulatory requirements, may result in
enforcement actions such as:
●
warning letters or
other actions requiring changes in product manufacturing processes
or restrictions on product marketing or distribution;
●
product recalls or
seizures or the temporary or permanent withdrawal of a product from
the market;
●
suspending any
ongoing clinical trials;
●
temporary or
permanent injunctions against our production
operations;
●
refusal of our
applications for pre-market approval or an investigational new drug
application; and
●
fines, restitution
or disgorgement of profits or revenue, the imposition of civil
penalties or criminal prosecution.
The
occurrence of any of these actions would likely cause a material
adverse effect on our business, financial condition and results of
operations.
Many of our business practices are subject to scrutiny and
potential investigation by regulatory and government enforcement
authorities, as well as to lawsuits brought by private citizens
under federal and state laws. We could become subject to
investigations, and our failure to comply with applicable law or an
adverse decision in lawsuits may result in adverse consequences to
us. If we fail to comply with U.S. healthcare laws, we could face
substantial penalties and financial exposure, and our business,
operations and financial condition could be adversely
affected.
While
payment is not yet available from third-party payors (government or
commercial) for our product, our goal is to obtain such coverage as
soon as possible after product approval and commercial launch in
the U.S . If this occurs, the availability of such payment would
mean that many healthcare laws would place limitations and
requirements on the manner in which we conduct our business
(including our sales and promotional activities and interactions
with healthcare professionals and facilities) and could result in
liability and exposure to us. In some instances, our interactions
with healthcare professionals and facilities that occurred prior to
commercialization could have implications at a later date. The laws
that may affect our ability to operate include, among others: (i)
the federal healthcare programs Anti-Kickback Statute, which
prohibits, among other things, persons from knowingly and willfully
soliciting, receiving, offering or paying remuneration, directly or
indirectly, in exchange for or to induce either the referral of an
individual for, or the purchase, order or recommendation of, any
good or service for which payment may be made under federal
healthcare programs such as Medicare or Medicaid, (ii) federal
false claims laws which prohibit, among other things, individuals
or entities from knowingly presenting, or causing to be presented,
claims for payment from Medicare, Medicaid, or other third-party
payors that are false or fraudulent, and which may apply to
entities like us under theories of “implied
certification” where the government and qui tam relators may
allege that device companies are liable where a product that was
paid for by the government in whole or in part was promoted
“off-label,” lacked necessary approval, or failed to
comply with good manufacturing practices or other laws; (iii)
transparency laws and related reporting and/or disclosures such as
the Sunshine Act; and/or (iv) state law equivalents of each of the
above federal laws, such as anti-kickback and false claims laws
which may apply to items or services reimbursed by any third-party
payor, including commercial insurers, many of which differ from
their federal counterparts in significant ways, thus complicating
compliance efforts.
If our
operations are found to be in violation of any of the laws
described above or any other governmental regulations that apply to
us, we may be subject to penalties, including civil and criminal
penalties, exclusion from participation in government healthcare
programs, damages, fines and the curtailment or restructuring of
our operations. Any penalties, damages, fines, curtailment or
restructuring of our operations could adversely affect our ability
to operate our business and our financial results. The risk of our
being found in violation of these laws is increased by the fact
that their provisions are open to a variety of evolving
interpretations and enforcement discretion. Any action against us
for violation of these laws, even if we successfully defend against
it, could cause us to incur significant legal expenses and divert
our management’s attention from the operation of our
business.
Both
federal and state government agencies have heightened civil and
criminal enforcement efforts. There are numerous ongoing
investigations of healthcare pharmaceutical companies and others in
the healthcare space, as well as their executives and managers. In
addition, amendments to the Federal False Claims Act, have made it
easier for private parties to bring qui tam (whistleblower)
lawsuits against companies under which the whistleblower may be
entitled to receive a percentage of any money paid to the
government. In addition, the Affordable Care Act amended the
federal civil False Claims Act to provide that a claim that
includes items or services resulting from a violation of the
federal anti-kickback statute constitutes a false or fraudulent
claim for purposes of the federal civil False Claims Act. Penalties
include substantial fines for each false claim, plus three times
the amount of damages that the federal government sustained because
of the act of that person or entity and/or exclusion from the
Medicare program. In addition, a majority of states have adopted
similar state whistleblower and false-claims provision. There can
be no assurance that our activities will not come under the
scrutiny of regulators and other government authorities or that our
practices will not be found to violate applicable laws, rules and
regulations or prompt lawsuits by private citizen "relators" under
federal or state false claims laws. Any future investigations of
our business or executives, or enforcement action or prosecution,
could cause us to incur substantial costs, and result in
significant liabilities or penalties, as well as damage to our
reputation.
Laws impacting the U.S. healthcare system are subject to a great
deal of uncertainty, which may result in adverse consequences to
our business.
There
have been a number of legislative and regulatory proposals to
change the healthcare system, reduce the costs of healthcare and
change medical reimbursement policies. Doctors, clinics, hospitals
and other users of our products may decline to purchase our
products to the extent there is uncertainty regarding coverage from
government or commercial payors. Further proposed legislation,
regulation and policy changes affecting third-party reimbursement
are likely. Among other things, Congress has in the past proposed
changes to and the repeal of the Patient Protection and Affordable
Care and Health Care and Education Affordability Reconciliation Act
of 2010 (collectively, the “Affordable Care Act”), and
lawsuits have been brought challenging aspects of the law at
various points. There have been repeated recent attempts by
Congress to repeal or replace the Affordable Care Act. At this
time, it remains unclear whether there will be any changes made to
or any repeal or replacement of the Affordable Care Act, with
respect to certain of its provisions or in its entirety. We are
unable to predict what legislation or regulation, if any, relating
to the health care industry or third-party coverage and
reimbursement may be enacted in the future at the state or federal
level, or what effect such legislation or regulation may have on
us. Denial of coverage and reimbursement of our products, or the
revocation or changes to coverage and reimbursement policies, could
have a material adverse effect on our business, results of
operations and financial condition.
We may not be successful in our efforts to build a pipeline of
product candidates.
A key
element of our strategy is to use and expand our product platform
to build a pipeline of product candidates, and progress those
product candidates through clinical development for the treatment
of a variety of different types of cancer. Even if we are
successful in building a product pipeline, the potential product
candidates that we identify may not be suitable for clinical
development for a number of reasons, including causing harmful side
effects or demonstrating other characteristics that indicate a low
likelihood of receiving marketing approval or achieving market
acceptance. If our methods of identifying potential product
candidates fail to produce a pipeline of potentially viable product
candidates, then our success as a business will be dependent on the
success of fewer potential product candidates, which introduces
risks to our business model and potential limitations to any
success we may achieve.
Our product candidates may cause undesirable side effects or have
other properties that could delay or prevent their regulatory
approval, limit the commercial profile of an approved label, or
result in significant negative consequences following marketing
approval, if any.
Additionally,
if one or more of our product candidates receives marketing
approval, and we or others later identify undesirable side effects
caused by such products, a number of potentially significant
negative consequences could result, including:
●
regulatory
authorities may withdraw approvals of such product;
●
regulatory
authorities may require additional warnings on the product’s
label;
●
we may be required
to create a medication guide for distribution to patients that
outlines the risks of such side effects;
●
we could be sued
and held liable for harm caused to patients; and
●
our reputation may
suffer.
Any of
these events could prevent us from achieving or maintaining market
acceptance of the particular product candidate, if approved, and
could significantly harm our business, results of operations and
prospects.
We may expend our limited resources to pursue a particular product
candidate or indication that does not produce any commercially
viable products and may fail to capitalize on product candidates or
indications that may be more profitable or for which there is a
greater likelihood of success.
Because
we have limited financial and managerial resources, we must focus
our efforts on particular research programs and product candidates
for specific indications. As a result, we may forego or delay
pursuit of opportunities with other product candidates or for other
indications that later prove to have greater commercial potential.
Further, our resource allocation decisions may result in our use of
funds for research and development programs and product candidates
for specific indications that may not yield any commercially viable
products. If we do not accurately evaluate the commercial potential
or target market for a particular product candidate, we may
relinquish valuable rights to that product candidate through
collaboration, licensing or other royalty arrangements in cases in
which it would have been more advantageous for us to retain sole
development and commercialization rights to such product candidate.
Any such failure to improperly assess potential product candidates
could result in missed opportunities and/or our focus on product
candidates with low market potential, which would harm our business
and financial condition.
Our products may be expensive to manufacture, and they may not be
profitable if we are unable to control the costs to manufacture
them.
Our
products may be significantly more expensive to manufacture than we
expect or than other therapeutic products currently on the market
today. We hope to substantially reduce manufacturing costs through
process improvements, development of new methods, increases in
manufacturing scale and outsourcing to experienced manufacturers.
If we are not able to make these, or other improvements, and
depending on the pricing of the product, our profit margins may be
significantly less than that of other therapeutic products on the
market today. In addition, we may not be able to charge a high
enough price for any product we develop, even if they are safe and
effective, to make a profit. If we are unable to realize
significant profits from our potential product candidates, our
business would be materially harmed.
For some of our products, we currently lack sufficient
manufacturing capabilities to produce our therapeutic product
candidates at commercial-scale quantities and do not have an
alternate manufacturing supply, which could negatively impact our
ability to meet any future demand for the product.
We
expect that we would need to significantly expand our manufacturing
capabilities to meet potential demand for our therapeutic product
candidates, if approved. Such expansion would require additional
regulatory approvals. Even if we increase our manufacturing
capabilities, it is possible that we may still lack sufficient
capacity to meet demand.
We do
not currently have any alternate supply for our products. If our
facilities where our products are currently being manufactured or
equipment were significantly damaged or destroyed, or if there were
other disruptions, delays or difficulties affecting manufacturing
capacity, including if such facilities are deemed not in compliance
with current Good Manufacturing Practice, or GMP, requirements,
future clinical studies and commercial production for our products
would likely be significantly disrupted and delayed. It would be
both time-consuming and expensive to replace this capacity with
third parties, particularly since any new facility would need to
comply with the regulatory requirements.
Ultimately,
if we are unable to supply our products to meet commercial demand,
whether because of processing constraints or other disruptions,
delays or difficulties that we experience, our production costs
could dramatically increase and sales of our products and their
long-term commercial prospects could be significantly
damaged.
To be successful, our proposed products must be accepted by the
healthcare community, which can be very slow to adopt or
unreceptive to new technologies and products.
Our
proposed products and those developed by our collaborative
partners, if approved for marketing, may not achieve market
acceptance since hospitals, physicians, patients or the medical
community in general may decide not to accept and use these
products. The products that we are attempting to develop represent
substantial departures from established treatment methods and will
compete with a number of more conventional therapies manufactured
and marketed by major pharmaceutical companies. The degree of
market acceptance of any of our developed products will depend on a
number of factors, including:
●
our establishment
and demonstration to the medical community of the clinical efficacy
and safety of our proposed products;
●
our ability to
create products that are superior to alternatives currently on the
market;
●
our ability to
establish in the medical community the potential advantage of our
treatments over alternative treatment methods; and
●
reimbursement
policies of government and third-party payers.
If the
healthcare community does not accept our products for any of these
reasons, or for any other reason, our business would be materially
harmed.
Our business is based on novel technologies that are inherently
expensive and risky and may not be understood by or accepted in the
marketplace, which could adversely affect our future
value.
The
clinical development, commercialization and marketing of
immuno-oncology therapies are at an early-stage, substantially
research-oriented, and financially speculative. To date, very few
companies have been successful in their efforts to develop and
commercialize an immuno-oncology therapeutic product. In general,
such products may be susceptible to various risks, including
undesirable and unintended side effects, unintended immune system
responses, inadequate therapeutic efficacy, or other
characteristics that may prevent or limit their approval or
commercial use. Furthermore, the number of people who may use such
therapies is difficult to forecast with accuracy. Our future
success is dependent on the establishment of a significant market
for such therapies and our ability to capture a share of this
market with our product candidates.
Our
development efforts with our therapeutic product candidates are
susceptible to the same risks of failure inherent in the
development and commercialization of therapeutic products based on
new technologies. The novel nature of immuno-oncology therapeutics
creates significant challenges in the areas of product development
and optimization, manufacturing, government regulation, third-party
reimbursement and market acceptance. For example, the FDA has
relatively limited experience regulating such therapies, and there
are few approved treatments using such therapy.
Our competition includes fully integrated biotechnology and
pharmaceutical companies that have significant advantages over
us.
The
market for therapeutic immuno-oncology products is highly
competitive. We expect that our most significant competitors will
be fully integrated and more established pharmaceutical and
biotechnology companies or institutions, including major
multinational pharmaceutical companies, biotechnology companies and
universities and other research institutions. These companies are
developing similar products, and they have significantly greater
capital resources and research and development, manufacturing,
testing, regulatory compliance, and marketing capabilities. Many of
these potential competitors may be further along in the process of
product development and also operate large, company-funded research
and development programs. As a result, our competitors may develop
more competitive or affordable products, or achieve earlier patent
protection or product commercialization than we are able to
achieve. Competitive products may render any products or product
candidates that we develop obsolete.
Many of
our competitors have substantially greater financial, technical and
other resources than we do, such as larger research and development
staff and experienced marketing and manufacturing organizations.
Additional mergers and acquisitions in the biotechnology and
pharmaceutical industries may result in even more resources being
concentrated in certain of our competitors. As a result, these
companies may be able to obtain regulatory approval more rapidly
than we can and may be more effective in selling and marketing
their products. Smaller or early-stage companies may also prove to
be significant competitors, particularly through collaborative
arrangements with large, established companies. Competition may
increase further as a result of advances in the commercial
applicability of technologies and greater availability of capital
for investment in these industries. Our competitors may succeed in
developing, acquiring or licensing drug products that are more
effective or less costly to produce or purchase on the market than
any product candidate we are currently developing or that we may
seek to develop in the future. If approved, our product candidates
will face competition from commercially available drugs as well as
drugs that are in the development pipelines of our
competitors.
Established
pharmaceutical companies may invest heavily to accelerate discovery
and development of or in-license novel compounds that could make
our product candidates less competitive. In addition, any new
product that competes with an approved product must demonstrate
compelling advantages in efficacy, convenience, tolerability and
safety in order to overcome price competition and to be
commercially successful. Accordingly, our competitors may succeed
in obtaining patent protection, receiving FDA, EMA or other
regulatory approval, or discovering, developing and commercializing
medicines before we do, which would have a material adverse impact
on our business and ability to achieve profitability from future
sales of our approved product candidates, if any.
If competitors develop and market products that are more effective,
safer or less expensive than our product candidates or offer other
advantages, our commercial prospects will be limited.
Our
therapeutic immuno-oncology development programs face, and will
continue to face, intense competition from pharmaceutical,
biopharmaceutical and biotechnology companies, as well as numerous
academic and research institutions and governmental agencies
engaged in drug discovery activities or funding, both in the United
States and abroad. Some of these competitors are pursuing the
development of drugs and other therapies that target the same
diseases and conditions that we are targeting with our product
candidates.
As a
general matter, we also face competition from many companies that
are researching and developing cell therapies. Many of these
companies have financial and other resources substantially greater
than ours. In addition, many of these competitors have
significantly greater experience in testing pharmaceutical and
other therapeutic products, obtaining FDA and other regulatory
approvals, and marketing and selling. If we ultimately obtain
regulatory approval for any of our product candidates, we also will
be competing with respect to manufacturing efficiency and marketing
capabilities, areas in which we have limited or no
commercial-scale experience. Mergers and acquisitions in the
pharmaceutical and biotechnology industries may result in even more
resources’ being concentrated by our competitors. Competition
may increase further as a result of advances made in the commercial
applicability of our technologies and greater availability of
capital for investment in these fields.
Our CNS
portfolio compounds also face considerable competition. Many
compounds are in development for the treatment of neuropathic pain.
Current treatments for neuropathic include narcotic analgesics,
voltage-gated sodium channel blockers, voltage-gated calcium
channel blockers, glutamate NMDA NR2B antagonists (ketamine), drugs
that increase monoamine transmission, and cannabinoids.
Some of the key players operating in
the global neuropathic pain market are Depomed Inc. (NASDAQ:DEPO),
Pfizer Inc. (NYSE:PFE), Johnson & Johnson (NYSE:JNJ),
Bristol-Myers Squibb (NYSE:BMY), Eli Lilly and Company (NYSE:LLY),
GlaxoSmithKline PLC (NYSE:GSK), Sanofi S.A. (NYSE:SNY), Biogen Idec
Inc. (NASDAQ:BIIB), and Baxter Healthcare Corporation (NYSE:BAX).
In the field of myasthenia gravis, pharmaceutical R&D efforts
focus on the discovery of a cure for the disease. A cure would make
treatment with GTP-004 obsolete. In the field of motion sickness,
research may be ongoing for better anti-motion sickness
drugs.
If we are unable to keep up with rapid technological changes in our
field or compete effectively, we will be unable to operate
profitably.
We are
engaged in activities in the biotechnology field, which is
characterized by extensive research efforts and rapid technological
progress. If we fail to anticipate or respond adequately to
technological developments, our ability to operate profitably could
suffer. Research and discoveries by other biotechnology,
agricultural, pharmaceutical or other companies may render our
technologies or potential products or services uneconomical or
result in products superior to those we develop. Similarly, any
technologies, products or services we develop may not be preferred
to any existing or newly developed technologies, products or
services.
We may not be able to obtain third-party patient reimbursement or
favorable product pricing, which would reduce our ability to
operate profitably.
Our
ability to successfully commercialize certain of our proposed
products in the human therapeutic field may depend to a significant
degree on patient reimbursement of the costs of such products and
related treatments at acceptable levels from government
authorities, private health insurers and other organizations, such
as health maintenance organizations. Reimbursement in the United
States or foreign countries may not be available for any products
we may develop, and, if available, may be decreased in the future.
Also, reimbursement amounts may reduce the demand for, or the price
of, our products with a consequent harm to our business. We cannot
predict what additional regulation or legislation relating to the
healthcare industry or third-party coverage and reimbursement may
be enacted in the future or what effect such regulation or
legislation may have on our business. If additional regulations are
overly onerous or expensive, or if healthcare-related legislation
makes our business more expensive or burdensome than originally
anticipated, we may be forced to significantly downsize our
business plans or completely abandon our business
model.
We may be subject to litigation that will be costly to defend or
pursue and uncertain in its outcome.
Our
business may bring us into conflict with our licensees, licensors
or others with whom we have contractual or other business
relationships, or with our competitors or others whose interests
differ from ours. If we are unable to resolve those conflicts on
terms that are satisfactory to all parties, we may become involved
in litigation brought by or against us. That litigation is likely
to be expensive and may require a significant amount of
management’s time and
attention, at the expense of other aspects of our business. The
outcome of litigation is always uncertain, and in some cases could
include judgments against us that require us to pay damages, enjoin
us from certain activities, or otherwise affect our legal or
contractual rights, which could have a significant adverse effect
on our business.
We are exposed to the risk of liability claims, for which we may
not have adequate insurance.
Since
we participate in the pharmaceutical industry, we may be subject to
liability claims by employees, customers, end users and third
parties. We do not currently have product liability insurance. We
intend to obtain proper insurance . however, there can be no
assurance that any liability insurance we purchase will be adequate
to cover claims asserted against us or that we will be able to
maintain such insurance in the future. We intend to adopt prudent
risk-management programs to reduce these risks and potential
liabilities. however, we have not taken any steps to create these
programs and have no estimate as to the cost or time required to do
so and there can be no assurance that such programs, if and when
adopted, will fully protect us. We may not be able to put risk
management programs in place, or obtain insurance, if we are unable
to retain the necessary expertise and/or are unsuccessful in
raising necessary capital in the future. Our failure to obtain
appropriate insurance, or to adopt and implement effective
risk-management programs, as well as any adverse rulings in any
legal matters, proceedings and other matters could have a material
adverse effect on our business.
Preclinical
and clinical trials are conducted during the development of
potential products and other treatments to determine their safety
and efficacy for use by humans. Notwithstanding these efforts, when
our treatments are introduced into the marketplace, unanticipated
side effects may become evident. Manufacturing, marketing, selling
and testing our product candidates under development or to be
acquired or licensed, entails a risk of product liability claims.
We could be subject to product liability claims in the event that
our product candidates, processes, or products under development
fail to perform as intended. Even unsuccessful claims could result
in the expenditure of funds in litigation and the diversion of
management time and resources, and could damage our reputation and
impair the marketability of our product candidates and processes.
While we plan to maintain liability insurance for product liability
claims, we may not be able to obtain or maintain such insurance at
a commercially reasonable cost. If a successful claim were made
against us, and we lacked insurance or the amount of insurance were
inadequate to cover the costs of defending against or paying such a
claim or the damages payable by us, we would experience a material
adverse effect on our business, financial condition and results of
operations.
We could be subject to product liability lawsuits based on the use
of our product candidates in clinical testing or, if obtained,
following marketing approval and commercialization. If product
liability lawsuits are brought against us, we may incur substantial
liabilities and may be required to cease clinical testing or limit
commercialization of our product candidates.
We
could be subject to product liability lawsuits if any product
candidate we develop allegedly causes injury or is found to be
otherwise unsuitable for human use during product testing,
manufacturing, marketing or sale. Any such product liability claims
may include allegations of defects in manufacturing, defects in
design, a failure to warn of dangers inherent in the product,
negligence, strict liability and a breach of warranties. Claims
could also be asserted under state consumer protection acts. If we
cannot successfully defend ourselves against product liability
claims, we may incur substantial liabilities or be required to
limit commercialization of our product candidates, if approved.
Even successful defense would require significant financial and
management resources. Regardless of the merits or eventual outcome,
liability claims may result in:
●
decreased demand
for our product candidates;
●
withdrawal of
clinical trial participants;
●
initiation of
investigations by regulators;
●
costs to defend the
related litigation;
●
a diversion of
management’s time and our resources;
●
substantial
monetary awards to trial participants or patients;
●
product recalls,
withdrawals or labeling, marketing or promotional
restrictions;
●
loss of revenues
from product sales; and
●
the inability to
commercialize our product candidates.
Our
inability to retain sufficient product liability insurance at an
acceptable cost to protect against potential product liability
claims could prevent or inhibit the clinical testing and
commercialization of products we develop. We may wish to obtain
additional such insurance covering studies or trials in other
countries should we seek to expand those clinical trials or
commence new clinical trials in other jurisdictions or increase the
number of patients in any clinical trials we may pursue. We also
may determine that additional types and amounts of coverage would
be desirable at later stages of clinical development of our product
candidates or upon commencing commercialization of any product
candidate that obtains required approvals. However, we may not be
able to obtain any such additional insurance coverage when needed
on acceptable terms or at all. If we do not obtain or retain
sufficient product liability insurance, we could be responsible for
some or all of the financial costs associated with a product
liability claim relating to our preclinical and clinical
development activities, in the event that any such claim results in
a court judgment or settlement in an amount or of a type that is
not covered, in whole or in part, by any insurance policies we may
have or that is in excess of the limits of our insurance coverage.
We may not have, or be able to obtain, sufficient capital to pay
any such amounts that may not be covered by our insurance
policies.
We rely on third parties to conduct preclinical and clinical trials
of our product candidates. If these third parties do not
successfully carry out their contractual duties or meet expected
deadlines, we may not be able to obtain regulatory approval for or
commercialize our product candidates and our business could be
substantially harmed.
We
rely, and expect to continue to rely, upon third-party CROs to
execute our preclinical and clinical trials and to monitor and
manage data produced by and relating to those trials. However, we
may not be able to establish arrangements with CROs when needed or
on terms that are acceptable to us, or at all, which could
negatively affect our development efforts with respect to our drug
product candidates and materially harm our business, operations and
prospects.
We will
have only limited control over the activities of the CRO we will
engaged to continue conduct our clinical trials including the
University of Minnesota for our phase 2 clinical trial for
OXS-1550. Nevertheless, we are responsible for ensuring that each
of our studies is conducted in accordance with the applicable
protocol, legal, regulatory and scientific standards, and our
reliance on any CRO does not relieve us of our regulatory
responsibilities. Based on our present expectations, we, our CROs
and our clinical trial sites are required to comply with good
clinical practices, or GCPs, for all of our product candidates in
clinical development. Regulatory authorities enforce GCPs through
periodic inspections of trial sponsors, principal investigators and
trial sites. If we or any of our CROs fail to comply with
applicable GCPs, the clinical data generated in the applicable
trial may be deemed unreliable and the FDA, EMA or comparable
foreign regulatory authorities may require us to perform additional
clinical trials before approving a product candidate for marketing,
which we may not have sufficient cash or other resources to support
and which would delay our ability to generate revenue from any
sales of such product candidate. In addition, our clinical trials
are required to be conducted with product produced in compliance
with current good manufacturing practice requirements, or cGMPs.
Our or our CROs’ failure to comply with those regulations may
require us to repeat clinical trials, which would also require
significant cash expenditures and delay the regulatory approval
process.
Agreements
governing relationships with CROs generally provide those CROs with
certain rights to terminate a clinical trial under specified
circumstances. If a CRO that we have engaged terminates its
relationship with us during the performance of a clinical trial, we
would be forced to seek an engagement with a substitute CRO, which
we may not be able to do on a timely basis or on commercially
reasonable terms, if at all, and the applicable trial would
experience delays or may not be completed. In addition, our CROs
are not our employees, and except for remedies available to us
under any agreements we enter with them, we are unable to control
whether or not they devote sufficient time and resources to our
clinical, nonclinical and preclinical programs. If CROs do not
successfully carry out their contractual duties or obligations or
meet expected deadlines, if they need to be replaced or if the
quality or accuracy of the clinical data they obtain is compromised
due to a failure to adhere to our clinical protocols, regulatory
requirements or for other reasons, our clinical trials may be
extended, delayed or terminated and we may not be able to obtain
regulatory approval for, or successfully commercialize, the
affected product candidates. As a result, our operations and the
commercial prospects for the effected product candidates would be
harmed, our costs could increase and our ability to generate
revenues could be delayed.
We contract with third parties for the supply of product candidates
for clinical testing and expect to contract with third parties for
the manufacturing of our product candidates for large-scale testing
and commercial supply. This reliance on third parties increases the
risk that we will not have sufficient quantities of our product
candidates or products or such quantities at an acceptable cost,
which could delay, prevent or impair our development or
commercialization efforts.
We
anticipate continuing our engagement of third parties to provide
our clinical supply as we advance our product candidates into and
through clinical development. We expect in the future to use third
parties for the manufacture of our product candidates for clinical
testing, as well as for commercial manufacture. We plan to enter
into long-term supply agreements with several manufacturers for
commercial supplies. We may be unable to reach agreement on
satisfactory terms with contract manufacturers to manufacture our
product candidates. Additionally, the facilities to manufacture our
product candidates must be the subject of a satisfactory inspection
before the FDA or other regulatory authorities approve a marketing
authorization for the product candidate manufactured at that
facility. We will depend on these third-party manufacturers for
compliance with the FDA’s and international regulatory
authority requirements for the manufacture of our finished
products. We do not control the manufacturing process of, and are
completely dependent on, our contract manufacturers for compliance
with cGMPs. If our manufacturers cannot successfully manufacture
material that conforms to our specifications and the FDA and other
regulatory authorities’ cGMP requirements, they will not be
able to secure and/or maintain regulatory approval for their
manufacturing facilities. In addition, we have no control over the
ability of our contract manufacturers to maintain adequate quality
control, quality assurance and qualified personnel. If the FDA or a
comparable foreign regulatory authority does not approve these
facilities for the manufacture of our product candidates or if it
withdraws any such approval in the future, we may need to find
alternative manufacturing facilities, which would significantly
impact our ability to develop, obtain regulatory approval for or
market our product candidates, if approved, and may subject us to
recalls or enforcement action for products already on the
market.
If any
of our product candidates are approved and contract manufacturers
fail to deliver the required commercial quantities of finished
product on a timely basis and at commercially reasonable prices,
and we are unable to find one or more replacement manufacturers
capable of production at a substantially equivalent cost, in
substantially equivalent volumes and quality and on a timely basis,
we would likely be unable to meet demand for our products and could
lose potential revenue. It may take several years to establish an
alternative source of supply for our product candidates and to have
any such new source approved by the FDA or any other relevant
regulatory authorities.
We currently have no marketing and sales force. If we are unable to
establish effective marketing and sales capabilities or enter into
agreements with third parties to market and sell our product
candidates, we may not be able to effectively market and sell our
product candidates, if approved, or generate product
revenues.
We
currently do not have a marketing or sales team for the marketing,
sales and distribution of any of our product candidates that are
able to obtain regulatory approval. In order to commercialize any
product candidates, we must build on a territory-by-territory basis
marketing, sales, distribution, managerial and other non-technical
capabilities or make arrangements with third parties to perform
these services, and we may not be successful in doing so. If our
product candidates receive regulatory approval, we intend to
establish an internal sales and marketing team with technical
expertise and supporting distribution capabilities to commercialize
our product candidates, which will be expensive and time consuming
and will require significant attention of our executive officers to
manage. Any failure or delay in the development of our internal
sales, marketing and distribution capabilities would adversely
impact the commercialization of any of our products that we obtain
approval to market. With respect to the commercialization of all or
certain of our product candidates, we may choose to collaborate,
either globally or on a territory-by-territory basis, with third
parties that have direct sales forces and established distribution
systems, either to augment our own sales force and distribution
systems or in lieu of our own sales force and distribution systems.
If we are unable to enter into such arrangements when needed on
acceptable terms or at all, we may not be able to successfully
commercialize any of our product candidates that receive regulatory
approval or any such commercialization may experience delays or
limitations. If we are not successful in commercializing our
product candidates, either on our own or through collaborations
with one or more third parties, our future product revenue will
suffer and we may incur significant additional losses.
Our business and operations would suffer in the event of system
failures.
Despite
the implementation of security measures, our internal computer
systems and those of our contractors and consultants are vulnerable
to damage from computer viruses, unauthorized access, natural
disasters, terrorism, war and telecommunication and electrical
failures. While we have not experienced any such system failure,
accident or security breach to date, if such an event were to occur
and cause interruptions in our operations, it could result in a
material disruption of our drug development programs. For example,
the loss of clinical trial data from completed or ongoing or
planned clinical trials could result in delays in our regulatory
approval efforts and we may incur substantial costs to attempt to
recover or reproduce the data. If any disruption or security breach
resulted in a loss of or damage to our data or applications, or
inappropriate disclosure of confidential or proprietary
information, we could incur liability and/or the further
development of our product candidates could be
delayed.
Our operations are vulnerable to interruption by natural disasters,
power loss, terrorist activity and other events beyond our control,
the occurrence of which could materially harm our
business.
Businesses
located in California have, in the past, been subject to electrical
blackouts as a result of a shortage of available electrical power,
and any future blackouts could disrupt our operations. We are
vulnerable to a major earthquake, wildfire and other natural
disasters, and we have not undertaken a systematic analysis of the
potential consequences to our business as a result of any such
natural disaster and do not have an applicable recovery plan in
place. We do not carry any business interruption insurance that
would compensate us for actual losses from interruption of our
business that may occur, and any losses or damages incurred by us
could cause our business to materially suffer.
We have not held regular annual meetings in the past, and if we are
required by the Delaware Court of Chancery to hold an annual
meeting pursuant to Section 211(c) of the Delaware General
Corporation Law, or the DGCL, it could result in the unanticipated
expenditure of funds, time and other Company
resources.
Section
2.2 of our bylaws provides that an annual meeting shall be held
each year on a date and at a time designated by our board of
directors, and Section 211(b) of the DGCL provides for an annual
meeting of stockholders to be held for the election of directors.
Section 211(c) of the DGCL provides that if there is a failure to
hold the annual meeting for a period of 13 months after the latest
to occur of the organization of the corporation, its last annual
meeting or last action by written consent to elect directors in
lieu of an annual meeting, the Delaware Court of Chancery may order
a meeting to be held upon the application of any stockholder or
director. Section 211(c) also provides that the failure to hold an
annual meeting shall not affect otherwise valid corporate acts or
result in a forfeiture or dissolution of the
corporation.
We have
not held regular annual meetings in the past because a substantial
majority of our stock is owned by a small number of stockholders,
making it easy to obtain written consent in lieu of a meeting when
necessary. In light of our historical liquidity constraints,
handling matters by written consent has allowed our Company to save
on the financial and administrative resources required to prepare
for and hold such annual meetings. To our knowledge, no stockholder
or director has requested our Company’s management to hold
such an annual meeting and no stockholder or director has applied
to the Delaware Court of Chancery seeking an order directing our
company to hold a meeting. However, if one or more stockholders or
directors were to apply to the Delaware Court of Chancery seeking
such an order, and if the Delaware Court of Chancery were to order
an annual meeting before we are prepared to hold one, the
preparation for the annual meeting and the meeting itself could
result in the unanticipated expenditure of funds, time, and other
Company resources.
Risks Related to Our Common Stock
There has been a limited public market for our common stock, and we
do not know whether one will develop to provide you adequate
liquidity. Furthermore, the trading price for our common stock,
should an active trading market develop, may be volatile and could
be subject to wide fluctuations in per-share price.
Our
common stock is listed for trading on the OTCQB under the trading
symbol “GTBP”; historically, however, there has
been a limited public market for our common stock. We cannot assure
you that an active trading market for our common stock will develop
or be sustained. The liquidity of any market for the shares of our
common stock will depend on a number of factors,
including:
●
the number of
stockholders;
●
our operating
performance and financial condition;
●
the market for
similar securities;
●
the extent of
coverage of us by securities or industry analysts; and
●
the interest of
securities dealers in making a market in the shares of our common
stock.
Even if
an active trading market develops, the market price for our common
stock may be highly volatile and could be subject to wide
fluctuations. In addition, the price of shares of our common stock
could decline significantly if our future operating results fail to
meet or exceed the expectations of market analysts and investors
and actual or anticipated variations in our quarterly operating
results could negatively affect our share price.
Other
factors may also contribute to volatility of the price of our
common stock and could subject our common stock to wide
fluctuations. These include, but are not limited to:
●
developments in the
financial markets and worldwide or regional economies;
●
announcements of
innovations or new products or services by us or our
competitors;
●
announcements by
the government relating to regulations that govern our
industry;
●
significant sales
of our common stock or other securities in the open
market;
●
variations in
interest rates;
●
changes in the
market valuations of other comparable companies; and
●
changes in
accounting principles.
Because our common stock may be deemed a low-priced
“penny” stock, an investment in our common stock should
be considered high-risk and subject to marketability
restrictions.
Historically,
the trading price of our common stock has been $5.00 per share or
lower, and deemed a penny stock, as defined in Rule 3a51-1 under
the Exchange Act, and subject to the penny stock rules of the
Exchange Act specified in rules 15g-1 through 15g-10. Those rules
require broker–dealers, before effecting transactions in any
penny stock, to:
●
deliver to the
customer, and obtain a written receipt for, a disclosure
document;
●
disclose certain
price information about the stock;
●
disclose the amount
of compensation received by the broker–dealer or any
associated person of the broker–dealer;
●
send monthly
statements to customers with market and price information about the
penny stock; and
●
in some
circumstances, approve the purchaser’s account under certain standards
and deliver written statements to the customer with information
specified in the rules.
Consequently,
if the price of our common stock returns to $5.00 per share or
lower, the penny stock rules may restrict the ability or
willingness of broker–dealers to sell the common stock and
may affect the ability of holders to sell their common stock in the
secondary market and the price at which such holders can sell any
such securities. These additional procedures could also limit our
ability to raise additional capital in the future.
If securities or industry analysts do not publish research or
reports about our business, or if they issue an adverse or
misleading opinion regarding our stock, our stock price and trading
volume could decline.
The
trading market for our common stock may be influenced by the
research and reports that industry or securities analysts publish
about us or our business. We do not currently have and may never
obtain research coverage by securities and industry analysts. If no
or few securities or industry analysts commence coverage of us, the
trading price for our common stock may be negatively affected. In
the event that we receive securities or industry analyst coverage,
if any of the analysts who cover us issue an adverse or misleading
opinion regarding us, our business model, our intellectual property
or our stock performance, or if our operating results fail to meet
the expectations of analysts, our stock price would likely decline.
If one or more of these analysts cease coverage of us or fail to
publish reports on us regularly, we could lose visibility in the
financial markets, which in turn could cause our stock price or
trading volume to decline.
Anti-takeover provisions may limit the ability of another party to
acquire us, which could cause our stock price to
decline.
Delaware
law and our charter, bylaws, and other governing documents contain
provisions that could discourage, delay or prevent a third party
from acquiring us, even if doing so may be beneficial to our
stockholders, which could cause our stock price to decline. In
addition, these provisions could limit the price investors would be
willing to pay in the future for shares of our common
stock.
We do not currently or for the foreseeable future intend to pay
dividends on our common stock.
We have
never declared or paid any cash dividends on our common stock. We
currently anticipate that we will retain future earnings for the
development, operation and expansion of our business and do not
anticipate declaring or paying any cash dividends for the
foreseeable future. As a result, any return on your investment in
our common stock will be limited to the appreciation in the price
of our common stock, if any.
ITEM
1B. UNRESOLVED STAFF
COMMENTS
Not
applicable.
ITEM
2. PROPERTIES
We
currently maintain offices at 1825 K Street NW, Suite 510,
Washington, D.C. 20006. We also maintained an office in Tampa,
Florida. Our lease for the Tampa office expired in December 2017
and was not renewed. Our total monthly
rent expense is approximately $9,000.
ITEM 3. LEGAL
PROCEEDINGS
On June 23, 2016, we were served with a complaint filed in the
Circuit Court of the 13th Judicial Circuit in and for Hillsborough
County, Florida, Case No. 16-CA-004791, by Lippert/Heilshorn and
Associates, Inc. Lippert/Heilshorn and Associates, Inc. is alleging
it is owed compensation for consulting services provided to us and
is seeking payment of $73,898. We have engaged legal counsel to
answer the complaint.
On February 15, 2017, MultiCell Immunotherapeutics, or MultiCell,
filed an arbitration proceeding against us with the American Health
Lawyers Association, Claim #3821. MultiCell is seeking
$207,783 plus interest and costs of arbitration pursuant to alleged
contract rights against us under a research agreement between
MultiCell and us. Following a hearing held September 1, 2017,
the arbitrator awarded MultiCell the payment amount of
$207,783 plus interest in the amount of $34,699. We have engaged
legal counsel to advise us in connection with this
matter.
ITEM
4. MINE SAFETY DISCLOSURES
None.
PART II
ITEM
5.
MARKET
FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS
AND ISSUER PURCHASES OF EQUITY SECURITIES.
Until
May 2009, our common stock was traded on the OTC Bulletin Board
(“OTCBB”) under the symbol “OXIS.” From May
20, 2009 until March 11, 2010, our common stock was traded on Pink
OTC Markets Inc. trading platform under the symbol
“OXIS.” From January 2015 to August 2017, our common
stock is quoted on the OTCQB under the “OXIS” trading
symbol. Since August 2017, our common stock is quoted on the OTCQB
under the “GTBP” trading symbol.
Trading
in our common stock has fluctuated greatly during the past year.
Accordingly, the prices for our common stock quoted on the OTCQB or
Pink OTC Markets Inc. may not necessarily be reliable indicators of
the value of our common stock. The following table sets forth the
high and low bid prices for shares of our common stock for the
quarters noted, as reported on the OTCQB and the Pink OTC Markets
Inc. The following price information reflects inter-dealer prices,
without retail mark-up, mark-down or commission and may not
represent actual transactions.
|
YEAR
|
|
PERIOD
|
|
HIGH
|
|
LOW
|
|
Fiscal
Year 2016
|
|
First
Quarter
|
|
960.00
|
|
123.00
|
|
|
|
Second
Quarter
|
|
180.00
|
|
93.00
|
|
|
|
Third
Quarter
|
|
108.00
|
|
45.00
|
|
|
|
Fourth
Quarter
|
|
57.00
|
|
11.79
|
|
Fiscal
Year 2017
|
|
First
Quarter
|
|
69.07
|
|
3.81
|
|
|
|
Second
Quarter
|
|
9.91
|
|
3.36
|
|
|
|
Third
Quarter
|
|
29.58
|
|
4.66
|
|
|
|
Fourth
Quarter
|
|
7.55
|
|
4.25
|
Our
common stock is also quoted on several European based exchanges
including Berlin (GTBP.BE), Frankfurt (GTBP.DE), the Euronext
(GTBP.NX) and Paris, (GTBP.PA). The foregoing trading prices
exclude trading on these foreign stock markets.
Stockholders
As of
December 31, 2017, there were 31 stockholders of record, which
total does not include stockholders who hold their shares in
“street name.” The transfer agent for our common stock
is ComputerShare, whose address is 350 Indiana Street, Golden, CO
80401.
Dividends
We have
not paid any dividends on our common stock to date and do not anticipate that we will pay
dividends in the foreseeable future. Any payment of cash dividends
on our common stock in the future will be dependent upon the amount
of funds legally available, our earnings, if any, our financial
condition, our anticipated capital requirements and other factors
that the Board of Directors may think are relevant. However, we
currently intend for the foreseeable future to follow a policy of
retaining all of our earnings, if any, to finance the development
and expansion of our business and, therefore, do not expect to pay
any dividends on our common stock in the foreseeable
future.
Equity Compensation Plan Information
The
information included under the heading “Equity Compensation
Plan Information” in Item 12 of Part III of this report,
“Security Ownership of Certain Beneficial Owners and
Management and Related Stockholder Matters.” is hereby
incorporated by reference into this Item 5 of this
report.
Recent Issuances of Unregistered Securities
We did
not issue any unregistered securities during the fourth quarter of
the fiscal year covered by this report.
Repurchase of Shares
We did
not repurchase any shares during the fourth quarter of the fiscal
year covered by this report.
ITEM 6. SELECTED FINANCIAL
DATA
This
company qualifies as a “smaller reporting company” as
defined in 17 C.F.R. §229.10(f)(1), and is not required to
provide information by this Item.
ITEM
7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
We are an immuno-oncology
company also focused on developing a portfolio of three central
nervous system, or CNS, product candidates. Our immuno-oncology
portfolio is based off a proprietary technology platform consisting
of single-chain bi-, tri- and tetra-specific scFv’s, combined
with proprietary antibody-drug linkers and drug payloads.
Constructs include bispecific and trispecific scFv constructs,
proprietary drug payloads, bispecific targeted antibody-drug
conjugates, or ADCs, as well as tri- and tetra-specific
antibody-directed cellular cytotoxicity, or ADCC. Our proprietary
tri- and tetra-specific ADCC platform engages natural killer cells,
or NK cells. NK cells are cytotoxic lymphocytes of the innate
immune system capable of immune surveillance. NK cells mediate ADCC
through the highly potent CD16 activating receptor. Upon
activation, NK cells deliver a store of membrane penetrating
apoptosis-inducing molecules. Unlike T cells, NK cells do not
require antigen priming.
Our CNS portfolio consists of innovative reformulations and/or
repurposing of existing therapies. These therapeutic agents address
certain unmet medical needs that can lead to improved efficacy
while addressing tolerability and safety issues that tended to
limit the usefulness of the original approved drug. Our CNS drug
candidates address disease states such as chronic neuropathic pain,
myasthenia gravis and vestibular disorders.
As
shown in the accompanying consolidated financial statements, the
Company has incurred an accumulated deficit of $267,896,000 through
December 31,
2017. On a consolidated basis, the Company had
cash and cash equivalents of $576,000 at December 31, 2017. Because our lack of
funds, we will have to raise additional capital in order to fund
our selling, general and administrative, and research and
development expenses. There are no assurances that we will be able
to raise the funds necessary to maintain our operations or to
implement our business plan. The consolidated financial statements
included in this Annual Report do not include any adjustments
relating to the recoverability and classification of recorded
assets, or the amounts and classification of liabilities that might
be necessary in the event we cannot continue our
operations.
Recent Developments
Amendment to Certificate of Incorporation
On July 17, 2017, we amended our Certificate of Incorporation for
the purpose of changing our name from Oxis International, Inc. to
GT Biopharma, Inc.
Agreement and Plan of Merger
On September 1, 2017, we entered into an Agreement and Plan of
Merger whereby we acquired 100% of the issued and outstanding
capital stock of GTP. GTP is a biotechnology company focused on
acquiring or discovering and patenting what it believes to be
close-to-market improved treatments for CNS disease (neurology and
pain) and shepherding the products through the FDA approval process
potentially to NDA. GTP products currently include a treatment for
neuropathic pain, the symptoms of myasthenia gravis, and motion
sickness. In exchange for the ownership of GTP, we issued a total
of 16,927,878 shares of our common stock to the three prior owners
of GTP, which represented 33% of our issued and outstanding capital
stock on a fully diluted basis at the time of closing.
Upon the consummation of the acquisition, Anthony J. Cataldo
resigned as our chief executive officer and was simultaneously
elected as executive chairman of the board of directors. Kathleen
Clarence-Smith, M.D., Ph.D., the founder of GTP, was then elected
as our chief executive officer and a member of the board of
directors.
As conditions to the acquisition of GTP, (i) we raised $4,540,000
upon the sale of debentures which were subsequently converted into
3,575,109 shares of restricted common stock and 208,224 shares of
Series J Preferred Stock to a total of nine persons or entities;
(ii) canceled debt in the amount $17,295,352 upon the issuance of
13,712,516 shares of common stock and 700,278 shares of Series J
Preferred Stock to a total of 26 persons or entities; (iii) issued
494,911 shares of common stock and 5,046 shares of Series J
Preferred Stock upon the cashless exercise of warrants to a total
of 22 persons or entities; and (iv) converted 25,000 shares of
Series H Preferred Stock and 1,666,667 Series I Preferred Stock
into 5,327,734 shares of common stock held by a total of three
persons or entities. All stock issuances were exempt from the
registration requirements of Section 5 of the Act of 1933, as
amended, or the Act, pursuant to Section 4(a)(2) of the Act because
the issuances did not involve any public offering.
Employment Contracts
In connection with the acquisition, we entered into employment
contracts on September 1, 2017, with Mr. Cataldo as executive
chairman, Dr. Clarence-Smith as chief executive officer, Dr.
Raymond Urbanski as chief medical officer and Steven Weldon as
chief financial officer. We entered into an employment contract on
November 15, 2017, with Mr. Cross as president and chief operating
officer.
On February 14, 2018, the Company entered into the First Amendment
to the Employment Agreement with Dr. Clarence-Smith, amending the
Employment Agreement, dated September 1, 2017, between the Company
and Dr. Clarence-Smith. Under the First Amendment, Dr.
Clarence-Smith’s title has been revised to reflect her new
position and she will be paid an annual salary of $500,000, paid in
equal monthly installment. All other terms of her original
Employment Agreement remain unchanged.
On February 14, 2018, the Company entered into a Consultant
Agreement with Mr. Cataldo. The term of the Consultant Agreement
lasts until August 31, 2020 and is terminable at will and is
subject to automatic extension for successive one-year periods. Mr.
Cataldo will be paid $41,666.67 per month during the term of the
Consultant Agreement and will be entitled to participate in the
Company’s bonus plans.
On February 15, 2018, the Company entered into an Executive
Employment Agreement with Mr. Cross, pursuant to which Mr. Cross
will be employed as the Company’s Chief Executive
Officer. The term of the Executive Employment Agreement
is three years and is terminable at will by either the Company or
Mr. Cross and subject to automatic extensions for successive one
year periods. Mr. Cross will be paid an annual salary of
$500,000, paid in equal monthly installment. Mr. Cross is also
entitled to participate in the Company’s bonus plans. Under
the Executive Employment Agreement, the Company has agreed that
it will recommend to the Board that the Company grant Mr.
Cross an option to purchase 2,000,000 shares of the Company’s
common stock at an exercise price equal to the fair market value of
each share as determined by the Board as of the date of the grant.
The stock option grant would vest according to the following
schedule: (i) 34% of the shares on February 15, 2018, (ii) 33% of
the shares on February 15, 2019, and (iii) 33% of the shares on
February 15, 2020.
TriKE Agreements
In
March 2017, we entered a new one-year Sponsored Research Agreement
with the University of Minnesota. The purpose of this agreement is
to determine toxicities and in vivo behavior in our TriKE technology, which we license from
the University of Minnesota.
In June
2017, we entered into a
co-development partnership agreement with Altor BioScience
Corporation in which the we will collaborate exclusively in the
clinical development of a novel 161533 TriKE fusion protein for
cancer therapies using our TriKE technology.
License Agreements
Pursuant
to a patent license agreement with the ID4, dated December 31,
2014, we received a non-exclusive, worldwide license to certain
intellectual property, including intellectual property related
to treating a p62-mediated
disease (e.g., multiple myeloma).
On
February 25, 2015, we licensed exclusive rights to
three antibody-drug
conjugates, or ADCs, that MCIT will prepare for further
evaluation by GTBP as prospective therapeutics for the treatment of
triple-negative breast cancer, and multiple myeloma and associated
osteolytic bone disease. Under the terms of the agreement, MCIT
will develop three ADC product candidates which contain
GTBP’s lead drug candidates OXS-2175 and
OXS-4235.
We executed an exclusive worldwide license agreement with Daniel A.
Vallera, Ph.D. and his associate (jointly "Dr. Vallera"), to
further develop and commercialize DT2219ARL (OXS-1550), a novel
therapy for the treatment of various human cancers. Under the terms
of the agreement, we receive exclusive rights to conduct research
and to develop, make, use, sell, and import DT2219ARL worldwide for
the treatment of any disease, state or condition in humans. GTBP
shall own all permits, licenses, authorizations, registrations and
regulatory approvals required or granted by any governmental
authority anywhere in the world that is responsible for the
regulation of products such as DT2219ARL, including without
limitation the FDA and the European Agency for the Evaluation of
Medicinal Products in the European Union. Under the agreement, Dr.
Vallera will receive an upfront license fee, royalty fees, and
certain milestone payments.
In
July 2016, we executed an exclusive worldwide license
agreement with the Regents of the University of Minnesota, to
further develop and commercialize cancer therapies using TriKE technology
developed by researchers at the university to target NK cells to
cancer. Under the terms of the agreement, we received
exclusive rights to conduct research and to develop, make, use,
sell, and import TriKe technology worldwide for the treatment of
any disease, state or condition in humans. We shall own all
permits, licenses, authorizations, registrations and regulatory
approvals required or granted by any governmental authority
anywhere in the world that is responsible for the regulation of
products such as the TriKe technology, including without limitation
the FDA and the European Agency for the Evaluation of Medicinal
Products in the European Union. Under the agreement, the University
of Minnesota will receive an upfront license fee, royalty fees, and
certain milestone payments.
In
September 2017, we in-licensed the rights to use the AccuBreak
patents with drugs that, like carbamazepine, are voltage-gated
sodium channel blockers in North America. The license field
includes voltage gated sodium channels inhibitors and blockers for
the treatment of epilepsy, neuropathic pain, and bipolar
disorder.
Financing
In
January 2016, the Company entered into a securities purchase
agreement with one accredited investor to sell 10% convertible
debentures, with and an exercise price of $1.25, with an initial
principal balance of $150,000 and warrants to acquire up to 80,000
shares of the Company's common stock at an exercise price of $1.25
per share.
In
May 2016, the Company entered into a securities purchase agreement
with twenty accredited investors to sell 10% convertible
debentures, with and an exercise price of $0.40, with an initial
principal balance of $1,390,044 and warrants to acquire up to
3,475,111 shares of the Company's common stock at an exercise price
of $0.45 per share.
In
July 2016, the Company entered into a securities purchase agreement
with one accredited investor to sell 10% convertible debentures,
with and an exercise price of $0.40, with an initial principal
balance of $112,135 and warrants to acquire up to 280,338 shares of
the Company's common stock at an exercise price of $0.45 per
share.
In
August 2016, the Company entered into a securities purchase
agreement with one accredited investor to sell 10% convertible
debentures up $1,000,000, with and an exercise price of $0.40, with
an initial principal balance of $250,000 and warrants to acquire up
to 2,500,000 shares of the Company's common stock at an exercise
price of $0.45 per share.
In
January 2017, the Company entered into a securities purchase
agreement with eight accredited investors to sell 10% convertible
debentures with and an exercise price of the lesser of (i) $15.00 or (ii) the
average of the three (3) lowest intra-day trading prices of the
Common Stock during the 20 Trading Days immediately prior to the
date on which the Notice of Conversion is delivered to the
Company, with an initial principal balance of $633,593
and warrants to acquire up to 42,239 shares of the Company's common
stock at an exercise price of $15.00 per share.
In
March 2017, the Company entered into a securities purchase
agreement with two accredited investors to sell 10% convertible
debentures with and an exercise price of the lesser of (i) $15.00 or (ii) the
average of the three (3) lowest intra-day trading prices of the
Common Stock during the 20 Trading Days immediately prior to the
date on which the Notice of Conversion is delivered to the
Company, with an initial principal balance of $232,313
and warrants to acquire up to 15,487 shares of the Company's common
stock at an exercise price of $15.00 per share.
In
April 2017, the Company entered into a securities purchase
agreement with two accredited investors to sell 10% convertible
debentures with and an exercise price of the lesser of (i) $15.00
or (ii) the average of the three (3) lowest intra-day trading
prices of the Common Stock during the 20 Trading Days immediately
prior to the date on which the Notice of Conversion is delivered to
the Company, with an initial principal balance of $70,000 and
warrants to acquire up to 4,666 shares of the Company's common
stock at an exercise price of $15.00 per share.
In
May 2017, the Company entered into a securities purchase agreement
with two accredited investors to sell 10% convertible debentures
with and an exercise price of the lesser of (i) $15.00 or (ii) the
average of the three (3) lowest intra-day trading prices of the
Common Stock during the 20 Trading Days immediately prior to the
date on which the Notice of Conversion is delivered to the Company,
with an initial principal balance of $125,000 and warrants to
acquire up to 8,333 shares of the Company's common stock at an
exercise price of $15.00 per share.
In July
2017, we entered into a securities purchase agreement with three
accredited investors to sell 10% convertible debentures with and an
exercise price of the lesser of (i) $15.00 or (ii) the average of
the three lowest intra-day trading prices of the common stock
during the 20 trading days immediately prior to the date on which
the notice of conversion is delivered to us, with an initial
principal balance of $650,000 and warrants to acquire up to 43,333
shares of our common stock at an exercise price of $15.00 per
share.
In
August 2017, we entered into a securities purchase agreement with
three accredited investors to sell 10% convertible debentures with
an exercise price of the lesser of (i) $15.00 or (ii) the average
of the three lowest intra-day trading prices of the common stock
during the 20 trading days immediately prior to the date on which
the notice of conversion is delivered to us, with an initial
principal balance of $3,890,000 and warrants to acquire up to
259,333 shares of our common stock at an exercise price of $15.00
per share.
On
January 22, 2018, the Company entered into a Securities Purchase
Agreement with the investors listed on the Schedule of Buyers
attached thereto (individually, a “Buyer” and
collectively, the “Buyers”) pursuant to which the
Company has agreed to issue to the Buyers senior convertible notes
in an aggregate principal amount of $7,760,510 (the
“Notes”), which Notes shall be convertible into the
Company’s common stock, par value $0.001 per share (the
“Common Stock”), and five-year warrants to purchase the
Company’s Common Stock representing the right to acquire an
aggregate of approximately 1,694,440 shares of Common Stock (the
“Warrants”).
The
issuance of the Notes and Warrants is being made in reliance on the
exemption provided by Section 4(a)(2) of the Securities Act of
1933, as amended (the “Securities Act”) for the offer
and sale of securities not involving a public offering, and
Regulation D promulgated under the Securities Act.
Contemporaneously
with the execution and delivery of the SPA, the Company and the
Buyers executed and delivered a Registration Rights Agreement (the
“Registration Rights Agreement”) pursuant to which the
Company has agreed to provide certain registration rights with
respect to the Registrable Securities (as defined thereto) under
the 1933 Act and the rules and regulations promulgated thereunder,
and applicable state securities laws. All descriptions of the SPA,
the Registration Rights Agreement, the Notes and the Warrants
contained herein are qualified in their entirety by reference to
the exhibits filed herewith.
Results of Operations
Research and Development
Expenses
During the year ended December 31, 2017 and 2016, we incurred
$1,068,000 and $975,000 of research and development
expenses.
Selling, general and administrative expenses
During the year ended December 31, 2017 and 2016, we incurred
$134,502,000 and $8,399,000 of selling, general and administrative
expenses. The increase in selling, general and
administrative expenses is primarily attributable to an increase of
non-cash stock compensation.
Change in value of warrant and derivative liabilities
During the year ended December 31, 2017, we recorded a gain as a
result of a decrease in the fair market value of outstanding
warrants and beneficial conversion features of
$925,000, compared to a gain of $25,697,000 during the year
ended December 31, 2016. We recorded a decrease as a result of the
conversion to common or preferred stock of all outstanding debt and
equity securities accounted for as derivative
liabilities.
Interest Expense
Interest expense was $8,602,000 and $6,555,000 for the year ended
December 31, 2017 and 2016 respectively. The decrease is
primarily due to an increase in the non-cash amortization of the
debt issuance costs associated with the convertible debentures and
demand notes payable and expenses related the issuance of
additional shares
Liquidity and Capital Resources
As of
December 31, 2017, we had cash and cash equivalents of $576,000.
This cash and cash equivalents is in part the result of the
proceeds from borrowings in 2017. On the same day we had total
current assets of $576,000, and a working capital deficit of
$2,103,000. Based upon the cash
position, it is necessary to raise additional capital by the end of
the next quarter in order to continue to fund current operations.
The Company is pursuing several alternatives to address this
situation, including the raising of additional funding through
equity or debt financings. In order to finance existing operations
and pay current liabilities over the next twelve months, the
Company will need to raise approximately $8-10 million of
capital.
Critical Accounting Policies
We
consider the following accounting policies to be critical given
they involve estimates and judgments made by management and are
important for our investors’ understanding of our operating
results and financial condition.
Basis of Consolidation
The
consolidated financial statements contained in this report include
the accounts of GT Biopharma, Inc. and its subsidiaries. All
intercompany balances and transactions have been
eliminated.
Revenue Recognition
License Revenue
License arrangements may consist of non-refundable upfront license
fees and various performance or sales milestones and future product
royalty payments. Some of these arrangements are
multiple element arrangements. Non-refundable, up-front
fees that are not contingent on any future performance by us, and
require no consequential continuing involvement on our part, are
recognized as revenue when the license term commences and the
licensed data, technology and/or compound is
delivered. We defer recognition of non-refundable
upfront fees if we have continuing performance obligations without
which the technology, right, product or service conveyed in
conjunction with the non-refundable fee has no utility to the
licensee that is separate and independent of our performance under
the other elements of the arrangement. In addition, if
we have continuing involvement through research and development
services that are required because our know-how and expertise
related to the technology is proprietary to us, or can only be
performed by us, then such up-front fees are deferred and
recognized over the period of continuing involvement.
Long-Lived
Assets
Our
long-lived assets include property, plant and equipment,
capitalized costs of filing patent applications and goodwill and
other assets. We evaluate our long-lived assets for impairment in
accordance with SFAS No. 144, “Accounting for the Impairment
or Disposal of Long-Lived Assets” whenever events or changes
in circumstances indicate that the carrying amount of such assets
may not be recoverable. Estimates of future cash flows and timing
of events for evaluating long-lived assets for impairment are based
upon management’s judgment. If any of our intangible or
long-lived assets are considered to be impaired, the amount of
impairment to be recognized is the excess of the carrying amount of
the assets over its fair value.
Applicable
long-lived assets are amortized or depreciated over the shorter of
their estimated useful lives, the estimated period that the assets
will generate revenue, or the statutory or contractual term in the
case of patents. Estimates of useful lives and periods of expected
revenue generation are reviewed periodically for appropriateness
and are based upon management’s judgment. Goodwill and other
assets are not amortized.
Certain Expenses and Liabilities
On an
ongoing basis, management evaluates its estimates related to
certain expenses and accrued liabilities. We base our estimates on
historical experience and on various other assumptions that we
believe to be reasonable under the circumstances, the results of
which form the basis for making judgments about the carrying values
of liabilities that are not readily apparent from other sources.
Actual results may differ materially from these estimates under
different assumptions or conditions.
Derivative Financial Instruments
During
the normal course of business, from time to time, we issue warrants
as part of a debt or equity financing. We do not enter into any
derivative contracts for speculative purposes. We recognize all
derivatives as assets or liabilities measured at fair value with
changes in fair value of derivatives reflected as current period
income or loss unless the derivatives qualify for hedge accounting
and are accounted for as such. During fiscal 2017 and 2016, we
issued warrants to purchase -0- and 5,101,500 shares of common
stock, respectively, in connection with equity transactions. In
accordance with ASC Topic 815-40, “Derivatives and Hedging
— Contracts in Entity’s Own Stock” (“ASC
815-40”), the value of these warrants is required to be
recorded as a liability, as the holders have an option to put the
warrants back to us in certain events, as defined.
Inflation
We
believe that inflation has not had a material adverse impact on our
business or operating results during the periods
presented.
Off-balance Sheet Arrangements
We have
no off-balance sheet arrangements as of December 31,
2017.
ITEM
7A QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT
MARKET RISK
This
company qualifies as a smaller reporting company, as defined in 17
C.F.R. §229.10(f) (1) and is not required to provide
information by this Item.
ITEM
8. FINANCIAL STATEMENTS AND SUPPLEMENTARY
DATA
Please
see the financial statements beginning on page F-1 located
elsewhere in this annual report and incorporated herein by
reference.
ITEM
9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS
ON ACCOUNTING AND FINANCIAL DISCLOSURE.
None.
ITEM
9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our
principal executive officer and principal financial officer
evaluated the effectiveness of our “disclosure controls and
procedures” (as such term is defined in Rules 13a-15(e) and
15d-15(e) of the United States Securities Exchange Act of 1934, as
amended), as of December 31, 2017. Based on that evaluation we have
concluded that our disclosure controls and procedures were not
effective as of December 31, 2017.
Management’s Report on Internal Control over Financial
Reporting
Management
is responsible for establishing and maintaining adequate internal
control over financial reporting. Internal control over financial
reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated
under the Securities Exchange Act of 1934, as amended, as a process
designed by, or under the supervision of, a company’s
principal executive and principal financial officers and effected
by a company’s board of directors, management and other
personnel, to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of financial
statements for external purposes in accordance with generally
accepted accounting principles and includes those policies and
procedures that:
●
Pertain to the
maintenance of records that in reasonable detail accurately and
fairly reflect the transactions and dispositions of the assets of
the company;
●
Provide reasonable
assurance that transactions are recorded as necessary to permit
preparation of financial statements in accordance with generally
accepted accounting principles, and that receipts and expenditures
of the company are being made only in accordance with
authorizations of management and directors of the company;
and
●
Provide reasonable
assurance regarding prevention or timely detection of unauthorized
acquisition, use or disposition of the company’s assets that
could have a material effect on the financial
statements.
All
internal control systems, no matter how well designed, have
inherent limitations and can provide only reasonable, not absolute,
assurance that the objectives of the control system are met.
Further, the design of a control system must reflect the fact that
there are resource constraints, and the benefits of controls must
be considered relative to their costs. Because of the inherent
limitations in all control systems, no evaluation of controls can
provide absolute assurance that all control issues and instances of
fraud, if any, within our company have been detected. Therefore,
even those systems determined to be effective can provide only
reasonable assurance with respect to financial statement
preparation and presentation.
As of
December 31, 2017, management of the company conducted an
assessment of the effectiveness of the company’s internal
control over financial reporting. In making this assessment, it
used the criteria set forth by the Committee of Sponsoring
Organizations of the Treadway Commission (COSO) in Internal
Control—Integrated Framework. In the course of the
assessment, material weaknesses were identified in the
company’s internal control over financial
reporting.
A
material weakness is a deficiency, or a combination of
deficiencies, in internal control over financial reporting, such
that there is a reasonable possibility that a material misstatement
of our annual or interim financial statements will not be prevented
or detected on a timely basis.
Management
determined that fundamental elements of an effective control
environment were missing or inadequate as of December 31, 2017. The
most significant issues identified were: 1) lack of segregation of
duties due to very small staff and significant reliance on outside
consultants, and 2) risks of executive override also due to lack of
established policies, and small employee staff. Based on the
material weaknesses identified above, management has concluded that
internal control over financial reporting was not effective as of
December 31, 2017.
As the
company’s operations increase, the company intends to hire
additional employees in its accounting department. This annual
report does not include an attestation report of our independent
registered public accounting firm regarding internal control over
financial reporting.
Changes in Internal Control over Financial Reporting
Other
than as described above, no changes in our internal control over
financial reporting were made during our most recent fiscal quarter
that has materially affected, or is reasonably likely to materially
affect, our internal control over financial reporting.
ITEM
9B. OTHER INFORMATION
None.
PART III
ITEM
10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE
GOVERNANCE
The
following table sets forth the name, age and position held by each
of our executive officers and directors as of February 20, 2018.
Directors are elected for a period of one year and thereafter serve
until the next annual meeting at which their successors are duly
elected by the stockholders.
|
Name
|
|
Age
|
|
Position
|
|
Shawn
Cross
|
|
50
|
|
Chief
Executive Officer and Chairman of the Board
|
|
Steven
Weldon
|
|
42
|
|
Chief
Financial Officer and Director
|
|
Raymond
Urbanski, M.D., Ph.D.
|
|
58
|
|
Chief
Medical Officer
|
|
Kathleen
Clarence-Smith, M.D., Ph.D.
|
|
71
|
|
Vice-Chairwoman
of the Board and President of Neurology Division
|
|
Anthony
J. Cataldo
|
|
66
|
|
Director
|
|
Geoffrey
Davis
|
|
70
|
|
Director
|
Shawn Cross has been
our President and Chief Operating Officer since November 2017 and
our Chief Executive Officer and Chairman of the Board since
February 15, 2018. Mr. Cross was a
Managing Director, and senior
calling officer focused on the biopharmaceutical industry, in
Healthcare Investment Banking at Deutsche Bank
Securities Inc. (NYSE:DB) from November 2015 to October 2017. He
was a Managing Director in
the Wells Fargo Securities,
LLC (NYSE:WF) Healthcare Group from December 2010 to November 2015.
Mr. Cross began his 20-year investment banking career at Alex.
Brown & Sons Inc. and has lived and worked in the major
financial centers of London, New York City and San
Francisco. He received his
bachelor of science degree from the University of California, Los
Angeles and his Master’s in Business Administration from
Columbia Business School with honors and a concentration in
Finance.
Steven Weldon was appointed to our board of directors in
September 2014 and as our Chief Financial Officer in November 2014.
Mr. Weldon has over 15 years of financial and accounting
experience. Mr. Weldon’s financial background includes
experience in managerial, private accounting and planning. He has
served on the board of several publicly traded companies as both,
chief executive officer and chief financial officer. Mr. Weldon was
appointed as chief financial officer and as a member of the board
of directors of GB Sciences, Inc. (OTCMKTS:GBLX) in September 2005
and served in both positions until November 2014. Mr. Weldon also
served as chief executive officer of GB Sciences from December
2009, through May 2011, and from April 2012, through March 2014.
For several years, he taught accounting and tax courses to
undergrad students at Florida Southern College. He received his
bachelor of science degree and his Master’s in Business
Administration from Florida Southern College and is a licensed
Certified Public Accountant in the State of Florida.
Raymond Urbanski, M.D., Ph.D., has been our Chief Medical Officer
since September 2017. Before joining us, he was the Chief Medical
Officer for MannKind Corporation (NASDAQ:MNKD) from August 2015 to
September 2017. He was the Chief Medical Officer for Mylan Inc.
(NASDAQ:MYL) from August 2012 to September 2014. Dr. Urbanski spent
eight years with Pfizer Inc. (NYSE:PFE), or Pfizer, and held
several positions with Pfizer, including Vice President and Chief
Medical Officer of the Established Products Business Unit, Senior
Medical Director of Oncology Clinical R&D, Senior Medical
Director of Breast Cancer Products and Medical Director of
Diversified Products. He brings extensive experience in developing
and overseeing clinical studies, including Phase 3b and Phase 4
studies (including line extensions) for sunitinib (Sutent),
exemestane (Aromasin), irinotecan (Camptosar), epirubicin
(Ellence), axitinib, IGF1R inhibitor, and tremilimumab. In addition
to his role with Pfizer, Dr. Urbanski has also served as chief
medical officer of Metabolex Inc. from October 2011 to June 2012,
and senior director of U.S. Medical Affairs for Aventis
(NYSE:SNY).
Anthony J. Cataldo served as our Executive Chairman since
September 1, 2017 to February 14, 2018. He was appointed to the
board of directors on July 31, 2014 and he served as Chief
Executive Officer from November 19, 2014 to September 1, 2017. Most
recently, from February 2011 to November 2013, Mr. Cataldo served
as Chairman and Chief Executive Officer of Iovance Biotherapeutics,
Inc. (NASDAQ: IOVA). Mr. Cataldo also served as chairman of the
board of directors of Brand Partners Group, Inc., a provider of
integrated products and services dedicated to providing financial
services and traditional retail clients with turnkey
environmental solutions, from October 2003 through August 2006. Mr.
Cataldo also served as nonexecutive cochairman of the
board of MultiCell Technologies, Inc., a supplier of functional,
nontumorigenic immortalized human hepatocytes from February
2005 through July 2006. Mr. Cataldo has also served as executive
chairman of Calypte Biomedical Corporation (OTCMKTS:CBMC), a
publicly traded biotechnology company, involved in the development
and sale of urine-based HIV1 screening tests from May 2001
through November 2004.
Kathleen Clarence-Smith, M.D., Ph.D., co-founded Georgetown
Translational Pharmaceuticals, Inc. (“GTP”) in
2015. Prior to
founding GTP, Dr. Clarence-Smith co-founded Chase Pharmaceuticals
Corporation in Washington D.C. and served as Chief Medical Officer
and member of the Chase Pharmaceuticals board from founding to
2014. Chase Pharmaceuticals was acquired by Allergan, PLC (NYSE:
AGN) in 2016. Dr. Clarence-Smith also held executive management
positions with Sanofi (NYSE:SNY), Roche Holding AG (VTX:ROG),
Otsuka Pharmaceutical, and Prestwick Scientific Capital. She is
co-founder and a managing member of KM Pharmaceutical Consulting in
Washington, D.C. She is currently serving as a member of the boards
of Riverside Pharmaceuticals Corporation, Westhaven Therapeutics
Corporation and Chase Therapeutics Corporation. Dr. Clarence-Smith
has been an observer of the board of the American Society for
Experimental Neurotherapeutics, or ASENT, since 1996 and has been a
member of the American Academy of Neurology since
1990.
Geoffrey Davis, is the founding
partner of Barker Davis LLC, a law firm specializing in advising
companies in the life sciences industry, and has been the managing
partner since its inception in January 2015. Prior to Barker Davis,
Mr. Davis was partner at Ropes & Gray LLC from September 1987
to December 2014. During his more than 25 years as a partner at
Ropes & Gray, Mr. Davis played a significant role in
establishing the firm’s internationally recognized Life
Sciences group. His work includes numerous corporate partnering and
licensing transactions for major pharmaceutical and medical device
companies, as well as for public and private biotechnology
companies and major medical centers. He has also worked extensively
on public and private financings for biomedical companies of all
sizes, ranging from newly organized companies to established
industry leaders, on behalf of both companies and their investment
bankers. He received his bachelor of arts degree from Yale
University and his Juris Doctor degree from Harvard Law
School.
Committees of the Board of Directors
Due to the small number of directors, at the present time the
duties of an Audit Committee, Nominating and Governance Committee,
and Compensation Committee are performed by the board of directors
as a whole. At such time as we have more directors on our board of
directors, these committees will be reconstituted.
Code of Ethics
A copy of the company’s code of ethics is attached to this
annual report as exhibit 99.
Section 16(a) Beneficial Ownership Reporting
Compliance
Section
16(a) of the Securities Exchange Act of 1934 requires our executive
officers and directors, and persons who own more than 10% of a
registered class of the company’s equity securities, to file
reports of ownership and changes in ownership with the Securities
and Exchange Commission (“SEC”). Executive officers,
directors and greater than 10% stockholders are required by SEC
regulations to furnish the company with copies of all Section 16(a)
forms they file. All of our executive officers and directors filed
the required reports; however, Kathleen Clarence-Smith and Raymond
Urbanski filed one Form 3 late and Anthony J. Cataldo and Steven
Weldon each filed one Form 4 late.
ITEM
11. EXECUTIVE COMPENSATION
The following table sets forth certain information concerning the
annual and long-term compensation for services rendered to us in
all capacities for the fiscal years ended December 31, 2017 and
2016 of all persons who served as our principal executive officers
and as our principal financial officer during the fiscal year ended
December 31, 2017. No other executive officers received total
annual compensation during the fiscal year ended December 31, 2017
in excess of $100,000. The principal executive officer and the
other named officers are collectively referred to as the
“Named Executive Officers.”
|
Name and
Principal Position
|
|
|
|
|
|
Non-Equity
Incentive Plan Compensation
($)
|
Nonqualified
Deferred Compensation Earnings
($)
|
All Other
Compensation
($)
|
|
|
Shawn
Cross(3)
|
2017
|
$104,165
|
$–––
|
$–––
|
$–––
|
$–––
|
$–––
|
$–––
|
$104,165
|
|
Chief Operating
Officer
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Steven
Weldon,
|
2017
|
$245,333
|
$–––
|
$38,472,797
|
$–––
|
$–––
|
$–––
|
$–––
|
$38,718,130
|
|
Chief Financial
Officer (Principal Financial Officer)(4)
|
2016
|
$168,000
|
$–––
|
$752,852
|
$–––
|
$–––
|
$–––
|
$–––
|
$920,852
|
|
|
2015
|
$168,000
|
$–––
|
$197,845
|
$–––
|
$–––
|
$–––
|
$–––
|
$365,845
|
|
|
|
|
|
|
|
|
|
|
|
Raymond Urbanski,
M.D., Ph.D.
|
2017
|
$133,333
|
$–––
|
$8,366,120
|
$–––
|
$–––
|
$–––
|
$–––
|
$8,499,453
|
|
Chief Medical
Officer
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Anthony J.
Cataldo,
|
2017
|
$310,667
|
$90,000
|
$77,275,253
|
$–––
|
$–––
|
$–––
|
$–––
|
$77,594,920
|
|
Former Chief
Executive Officer(5)
|
2016
|
$216,000
|
$–––
|
$4,417,026
|
$20,707
|
$–––
|
$–––
|
$–––
|
$4,653,733
|
|
|
2015
|
$216,000
|
$134,000
|
$–––
|
$102,535
|
$–––
|
$–––
|
$–––
|
$452,535
|
|
|
|
|
|
|
|
|
|
|
|
Kathleen
Clarence-Smith, M.D., Ph.D.(6)
|
2017
|
166,667
|
$–––
|
$–––
|
$–––
|
$–––
|
$–––
|
$–––
|
166,667
|
|
Former Chief
Executive Officer
|
|
|
|
|
|
|
|
|
|
(1)
The amounts in this column represent the aggregate
grant date fair value of the restricted stock awards and restricted
stock units, determined in accordance with Financial Accounting
Standards Board (FASB) Accounting Standards Codification (ASC)
Topic 718. GT Biopharma determines the grant date fair value of the
awards by multiplying the number of units granted by the closing
market price of one share of GT Biopharma common stock on the award
grant date. These amounts do not
reflect the actual economic value that will be realized by the
named executive officer upon the vesting or the sale of the common
stock awards.
(2)
This column
represents option awards computed in accordance with FASB ASC Topic
718, excluding the effect of estimated forfeitures related to
service-based vesting conditions. For additional information on the
valuation assumptions with respect to the option grants, refer to
Note 1 of our financial statements in this Annual Report. These
amounts do not correspond to the actual value that will be
recognized by the named executives from these awards.
(3)
Mr. Cross was
appointed President and Chief Operating Officer on October 15, 2017
and Chairman and Chief Executive Officer on February 14,
2018.
(4)
Mr. Weldon was
appointed Chief Financial Officer on November 3, 2014.
(5)
Dr. Urbanski was
appointed Chief Medical Officer on September 1, 2017.
(6)
Mr. Cataldo
served as our Chief Executive Officer from March 2009 to August
2011 and again in November 2014 to September 1, 2017. He was
Executive Chairman from September 1, 2017 to February 14,
2018.
(7)
Dr. Clarence-Smith
was Chief Executive Officer on September 1, 2017 to February 14,
2018. Dr. Clarence-Smith currently serves as our Vice-Chairwoman
and President of the Neurology Division.
Employment Agreements
We entered into employment contracts with our executive officers on
September 1, 2017, with Mr. Cataldo as executive chairman, Dr.
Clarence-Smith as chief executive officer, Dr. Urbanski as chief
medical officer and Mr. Weldon as chief financial
officer.
Mr. Cataldo’s contract is for three years. Under the terms of
his contract, he received an up-front restricted stock award of
5,129,600 common shares and will be paid an annual salary of
$500,000. Dr. Clarence-Smith’s contract is for three years.
Under the terms of her contract, she will receive an annual salary
of $500,000 and an up-front restricted stock award in an amount to
be determined by our board. Dr. Urbanski’s contract is for
three years. Under the terms of his contract, he received a
restricted stock award of 1,528,898 common shares that vest over two years
and will be paid an annual salary of $400,000. Mr. Weldon’s
contract is for three years pursuant to which he received an
up-front restricted stock award of 2,564,830 common shares and will
be paid an annual salary of $400,000. All four executives are
entitled to participate in any performance business plan
established by us.
On February 14, 2018, the Company entered into the First Amendment
to the Employment Agreement with Dr. Clarence-Smith, amending the
Employment Agreement, dated September 1, 2017, between the Company
and Dr. Clarence-Smith. Under the First Amendment, Dr.
Clarence-Smith’s title has been revised to reflect her new
position and she will be paid an annual salary of $500,000, paid in
equal monthly installment. All other terms of her original
Employment Agreement remain unchanged.
On February 14, 2018, the Company entered into a Consultant
Agreement with Mr. Cataldo. The term of the Consultant Agreement
lasts until August 31, 2020 and is terminable at will and is
subject to automatic extension for successive one-year periods. Mr.
Cataldo will be paid $41,666.67 per month during the term of the
Consultant Agreement and will be entitled to participate in the
Company’s bonus plans.
In November 16, 2017, Shawn Cross was appointed the President and
Chief Operating Officer of the GT Biopharma, Inc. by the Board of
Directors. The Company has entered into an Employment
agreement with Mr. Cross. Mr. Cross’s contract is
for three years and under the terms of his contract, he will
receive restricted stock awards of 150,000 common shares on January
1, 2018, 1,000,000 common shares on January 1, 2019 and 1,850,000
common shares on January 1, 2020 and will be paid an annual salary
of $500,000. He is also entitled to participate in any performance
business plan established by the Company.
On February 15, 2018, the Company entered into an Executive
Employment Agreement with Mr. Cross, pursuant to which Mr. Cross
will be employed as the Company’s Chief Executive
Officer. The term of the Executive Employment Agreement
is three years and is terminable at will by either the Company or
Mr. Cross and subject to automatic extensions for successive one
year periods. Mr. Cross will be paid an annual salary of
$500,000, paid in equal monthly installment. Mr. Cross is also
entitled to participate in the Company’s bonus plans. Under
the Executive Employment Agreement, the Company has agreed that
it will recommend to the Board that the Company grant Mr.
Cross an option to purchase 2,000,000 shares of the Company’s
common stock at an exercise price equal to the fair market value of
each share as determined by the Board as of the date of the grant.
The stock option grant would vest according to the following
schedule: (i) 34% of the shares on February 15, 2018, (ii) 33% of
the shares on February 15, 2019, and (iii) 33% of the shares on
February 15, 2020.
If any
of our executive officers’ employment with us is terminated
involuntarily, or any executive resigns with good reason as a
result of a change in control, the executive will receive (i) all
compensation and benefits earned through the date of termination of
employment; (ii) a lump-sum payment equal to the greater of (a) the
bonus paid or payable to the executive for the year immediately
prior to the year in which the change in control occurred and (b)
the target bonus under the performance bonus plan in effect
immediately prior to the year in which the change in control
occurs; (iii) a lump-sum payment equivalent to the remaining base
salary (as it was in effect immediately prior to the change in
control) due to the executive from the date of involuntary
termination to the end of the term of the employment agreement or
one half of the executive’s base salary then in effect,
whichever is the greater; and (iv) reimbursement for the cost
of medical, life, disability insurance coverage at a level
equivalent to that provided by us for a period expiring upon the
earlier of (a) one year or (b) the time the executive begins
alternative employment where said insurance coverage is available
and offered to the executive.
Stock Option Grants
The
following table sets forth information as of December 31, 2017,
concerning unexercised options, unvested stock and equity incentive
plan awards for the executive officers named in the Summary
Compensation Table.
|
|
|
|
Name
|
Number of
Securities Underlying Unexercised Options (#)
Exercisable
|
Number of
Securities Underlying Unexercised Options (#)
Unexercisable
|
Equity
Incentive Plan Awards: Number of Securities Underlying Unexercised
Unearned Options (#)
|
|
Option
Expiration Date
|
|
|
|
|
|
|
|
|
Anthony
Cataldo
|
358
|
-
|
-
|
$750.00
|
07/01/19
|
|
Anthony
Cataldo
|
358
|
-
|
-
|
$1,500.00
|
07/01/19
|
|
Anthony
Cataldo
|
358
|
-
|
-
|
$2,250.00
|
07/01/19
|
|
Steven
Weldon
|
-
|
-
|
-
|
-
|
|
|
Kathleen
Clarence-Smith, M.D., Ph.D.
|
-
|
-
|
-
|
-
|
|
|
Raymond Urbanski,
M.D., Ph.D.
|
-
|
-
|
-
|
-
|
|
|
Shawn
Cross
|
-
|
-
|
-
|
-
|
|
Director Compensation
Beginning
in January 2018, non-employee members of the Board of Directors are
to receive $42,500 per year, plus $15,000 annually for Chairing a
Committee and $5,000 annually as a member of a Committee. Also,
Directors are granted 150,000 options that vest over a three year
period. Vesting will accelerate if the Company undergoes a change
of control transaction for cash. There was not compensation paid to
non-employee directors during fiscal 2017.
ITEM
12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL
OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information regarding
beneficial ownership of our common stock as of February 20, 2018,
(a) by each person known by us to own beneficially 5% or more of
any class of our common stock, (b) by each of our named executive
officers, (c) by each of our directors and (d) by all our current
executive officers and directors as a group. As of February 20,
2018, there were 50,117,978 shares of our common stock issued and
outstanding. Shares of common stock subject to stock options and
preferred stock that are currently exercisable or exercisable
within 60 days of February 20, 2018 are deemed to be outstanding
for computing the percentage ownership of that person but are not
treated as outstanding for computing the percentage ownership of
any other person. Unless indicated below, the persons and entities
named in the table have sole voting and sole investment power with
respect to all shares beneficially owned, subject to community
property laws where applicable. Except as otherwise indicated, the
address of each stockholder is c/o GT Biopharma, Inc. at 1825 K
Street NW, Suite 510, Washington, D.C. 20006.
|
Name and
Address of Beneficial Owner
|
Number of
Shares of Common Stock Beneficially Owned
|
Percent of
Shares of Outstanding Common Stock
|
|
Security
Ownership of Certain Beneficial Owners:
|
|
|
|
Mark
Silverman
|
8,172,079
|
16.31%
|
|
Bristol Investment
Fund, Ltd. (1)
|
4,060,185
|
9.62%
|
|
Theorem Group, LLC
(2)
|
3,540,130
|
7.06%
|
|
William
Heavener
|
2,584,568
|
5.16%
|
|
Canyons
Trust
|
2,560,822
|
5.11%
|
|
Adam
Kasower
|
2,514,258
|
5.02%
|
|
Security
Ownership of Management:
|
|
|
|
Kathleen
Clarence-Smith, M.D., Ph.D.
|
8,505,633
|
16.97%
|
|
Anthony J.
Cataldo
|
5,143,036
|
10.26%
|
|
Steven
Weldon
|
2,566,835
|
5.12%
|
|
Raymond Urbanski,
M.D., Ph.D.
|
1,528,898
|
3.05%
|
|
Shawn
Cross
|
-
|
0.00%
|
|
|
|
|
|
Executive officers
and directors as a group — 6 persons
|
17,744,402
|
35.41%
|
(1)
As reported on Schedule 13G/A filed with the SEC on February 7,
2018. Paul Kessler, manager of Bristol Capital Advisors, LLC, the
investment advisor to Bristol Investment Fund, Ltd., has voting and
investment control over the securities held by Bristol Investment
Fund, Ltd. Mr. Kessler disclaims beneficial ownership of these
securities except to the extent of his pecuniary interest therein.
The address of Bristol Capital Advisors, LLC is 662 N. Sepulveda
Blvd., Suite 300, Los Angeles, California 90049.
(2)
As reported on Schedule 13G filed with the SEC on November 14,
2017. The address of Theorem Group LLC is 315 Beverly Drive, Suite
502, Beverly Hills, CA 90212
Equity Compensation Plan Information
The
following is a summary of our equity compensation plans at December
31, 2017:
|
Plan
Category
|
Number of
Securities to be Issued Upon Exercise of Outstanding Options,
Warrants and Rights
(a)
|
Weighted-Average
Exercise Price of Outstanding Options, Warrants and
Rights
(b)
|
Number of
Securities Remaining Available for Future Issuance Under Equity
Compensation Plans (Excluding Securities Reflected in Column
(a))
(c)
|
|
Equity compensation
plans approved by security holders (1)
|
1,246
|
$1,428.00
|
-
|
|
Equity compensation
plans not approved by security holders
|
-
|
-
|
-
|
|
Total
|
1,246
|
$1,428.00
|
-
|
(1)
As of December 31,
2017, we had options issued and outstanding to purchase 1,246
shares of common stock under our 2014 Stock Incentive
Plan.
ITEM
13. CERTAIN RELATIONSHIPS AND RELATED
TRANSACTIONS AND DIRECTOR INDEPENDENCE
Director Independence
One of
our four directors qualify as “independent directors”
as defined by Item 407 of Regulation S-K.
We have
elected to use the definition for “director
independence” under the Nasdaq Stock Market’s listing
standards, which defines an “independent director” as
“a person other than an officer or employee of us or its
subsidiaries or any other individual having a relationship, which
in the opinion of our Board of Directors, would interfere with the
exercise of independent judgment in carrying out the
responsibilities of a director.” The definition further
provides that, among others, employment of a director by us (or any
parent or subsidiary of ours) at any time during the past three
years is considered a bar to independence regardless of the
determination of our Board of Directors.
ITEM
14. PRINCIPAL ACCOUNTANT FEES AND
SERVICES
Seligson
& Giannattasio, LLP was our independent registered public
accounting firm for the fiscal years ending December 31, 2016 and
2017. The Audit Committee appointed Seligson & Giannattasio,
LLP as our independent registered public accounting firm for the
fiscal year ending December 31, 2017. The following table shows the
fees that were paid or accrued by us for audit and other services
provided by Seligson & Giannattasio, LLP for the 2016 and 2017
fiscal years.
|
|
|
|
|
Audit Fees
(1)
|
$56,000
|
$56,000
|
|
Audit-Related Fees
(2)
|
-
|
-
|
|
Tax Fees
(3)
|
4,000
|
4,000
|
|
All Other
Fees
|
-
|
-
|
|
Total
|
$60,000
|
$60,000
|
_____________
(1)
Audit fees
represent fees for professional services provided in connection
with the audit of our annual financial statements and the review of
our financial statements included in our Form 10-Q quarterly
reports and services that are normally provided in connection with
statutory or regulatory filings for the 2017 and 2016 fiscal
years.
(2)
Audit-related fees
represent fees for assurance and related services that are
reasonably related to the performance of the audit or review of our
financial statements and not reported above under “Audit
Fees.”
(3)
Tax fees represent
fees for professional services related to tax compliance, tax
advice and tax planning.
All
audit related services, tax services and other services rendered by
Seligson & Giannattasio, LLP were pre-approved by our Board of
Directors or Audit Committee. The Audit Committee has adopted a
pre-approval policy that provides for the pre-approval of all
services performed for us by Seligson & Giannattasio, LLP. The
policy authorizes the Audit Committee to delegate to one or more of
its members pre-approval authority with respect to permitted
services. Pursuant to this policy, the Board delegated such
authority to the Chairman of the Audit Committee. All pre-approval
decisions must be reported to the Audit Committee at its next
meeting. The Audit Committee has concluded that the provision of
the non-audit services listed above is compatible with maintaining
the independence Seligson & Giannattasio, LLP.
PART IV
ITEM
15. EXHIBITS, FINANCIAL STATEMENT
SCHEDULES
The
Company’s financial statements and related notes thereto are
listed and included in this Annual Report beginning on page F-1.
The following documents are furnished as exhibits to this Annual
Report on Form 10-K.
EXHIBIT
INDEX
|
|
|
|
|
Incorporated
by Reference
|
|
Exhibit
Number
|
|
Exhibit
Description
|
|
Form
|
|
Date
|
|
Number
|
|
Filed
Herewith
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Agreement
and Plan of Merger
|
|
10-Q
|
|
11/14/17
|
|
2.1
|
|
|
|
|
|
Restated
Certificate of Incorporation as filed in Delaware September 10,
1996 and as thereafter amended through March 1, 2002
|
|
10-KSB
|
|
04/01/02
|
|
3.A
|
|
|
|
|
|
Certificate
of Amendment to Amended and Restated Certificate of Incorporation
of GT Biopharma, Inc.
|
|
10-K
|
|
03/31/11
|
|
3.2
|
|
|
|
|
|
Certificate
of Designation of Preferences, Rights and Limitations of
Series H Convertible Preferred Stock of GT Biopharma, Inc.,
dated February 5, 2010
|
|
8-K
|
|
02/16/10
|
|
3.1
|
|
|
|
|
|
Certificate
of Designation of Preferences, Rights and Limitations of
Series I Convertible Preferred Stock of GT Biopharma, Inc.,
dated March 18, 2011.
|
|
10-K
|
|
03/31/11
|
|
3.4
|
|
|
|
|
|
Bylaws,
as restated effective September 7, 1994 and as amended through
April 29, 2003
|
|
10-QSB
|
|
08/13/03
|
|
3
|
|
|
|
|
|
License
Agreement with ID4 Pharma LLC
|
|
10-Q
|
|
08/11/17
|
|
10.1
|
|
|
|
|
|
License
Agreement with MultiCell Immunotherapeutics, Inc.
|
|
10-Q
|
|
08/11/17
|
|
10.2
|
|
|
|
|
|
License
Agreement with the University of Minnesota
|
|
10-Q
|
|
08/11/17
|
|
10.3
|
|
|
|
|
|
License
Agreement with Daniel A. Vallera, Ph.D.
|
|
10-Q
|
|
08/11/17
|
|
10.4
|
|
|
|
|
|
Employment
Agreement with Anthony Cataldo
|
|
10-Q
|
|
11/14/17
|
|
10.1
|
|
|
|
|
|
Employment
Agreement with Dr. Kathleen Clarence-Smith
|
|
10-Q
|
|
11/14/17
|
|
10.3
|
|
|
|
|
|
Employment
Agreement with Steven Weldon
|
|
10-Q
|
|
11/14/17
|
|
10.4
|
|
|
|
|
|
Employment
Agreement with Dr. Raymond Urbanski
|
|
10-Q
|
|
11/14/17
|
|
10.5
|
|
|
|
|
|
Warrant
Conversion Agreement
|
|
10-Q
|
|
11/14/17
|
|
10.6
|
|
|
|
|
|
Preferred
Conversion Agreement
|
|
10-Q
|
|
11/14/17
|
|
10.7
|
|
|
|
|
|
Amended
Note Conversion Agreement
|
|
10-Q
|
|
11/14/17
|
|
10.8
|
|
|
|
|
|
Amended
Warrant Conversion Agreement
|
|
10-Q
|
|
11/14/17
|
|
10.9
|
|
|
|
|
|
Amended
Preferred Conversion Agreement
|
|
10-Q
|
|
11/14/17
|
|
10.10
|
|
|
|
|
|
Agreement,
effective as of November 16, 2017, between the Company and Mr.
Cross
|
|
8-K
|
|
11/16/17
|
|
10.1
|
|
|
|
|
|
Securities
Purchase Agreement
|
|
8-K
|
|
01/13/17
|
|
10.1
|
|
|
|
|
|
10%
Senior Convertible Debenture
|
|
8-K
|
|
01/13/17
|
|
10.2
|
|
|
|
|
|
Common
Stock Purchase Warrant
|
|
8-K
|
|
01/13/17
|
|
10.3
|
|
|
|
|
|
Executive
Employment Agreement, dated as of February 15, 2018, between the
Company and Cross
|
|
8-K
|
|
02/21/18
|
|
10.1
|
|
|
|
|
|
First
Amendment to the Employment Agreement, dated as of February 14,
2018, between the Company and Dr. Clarence-Smith
|
|
8-K
|
|
02/21/18
|
|
10.2
|
|
|
|
|
|
Consulting
Agreement, dated as of February 14, 2018, between the Company and
Mr. Cataldo
|
|
8-K
|
|
02/21/18
|
|
10.3
|
|
|
|
|
|
Code of
Ethics
|
|
10-K
|
|
03/31/16
|
|
14.1
|
|
|
|
|
|
Subsidiaries
of GT Biopharma, Inc.
|
|
10-K
|
|
03/31/16
|
|
21.1
|
|
|
|
|
|
Certification
of the Principal Executive Officer pursuant to Rule
13a-14(a)/15d-14(a), as adopted pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
|
|
Certification
of the Principal Financial Officer pursuant to Rule
13a-14(a)/15d-14(a), as adopted pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
|
|
Certification
of the Principal Executive Officer pursuant to 18 U.S.C. Section
1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act
of 2002.
|
|
|
|
|
|
|
|
X
|
|
|
|
Certification
of the Principal Financial Officer pursuant to 18 U.S.C. Section
1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act
of 2002.
|
|
|
|
|
|
|
|
X
|
|
101
|
|
Interactive
Data File
|
|
|
|
|
|
|
|
X
|
SIGNATURES
Pursuant to the
requirements of Section 13 or 15(d) of the Securities Exchange Act
of 1934, the registrant has duly caused this report to be signed on
its behalf by the undersigned, thereunto duly
authorized.
|
Dated:
March 1, 2018
|
GT
Biopharma, Inc.
By:
/s/ Shawn
Cross
Shawn
Cross
Chief
Executive Officer and Chairman of the Board
|
|
|
|
Pursuant to the
requirements of the Securities Exchange Act of 1934, this report
has been signed below by the following persons on behalf of the
registrant and in the capacities and on the dates
indicated.
|
Name
|
|
Position
|
|
Date
|
|
|
|
|
|
|
|
/s/ Shawn Cross
Shawn
Cross
|
|
Chief
Executive Officer and Chairman of the Board
|
|
March
1, 2018
|
|
/s/ Steven Weldon
Steven
Weldon
|
|
Chief
Financial Officer (Principal Financial Officer), and
Director
|
|
March
1, 2018
|
|
/s/ Dr. Kathleen Clarence-Smith
Dr.
Kathleen Clarence-Smith
|
|
Vice
Chairwoman and Director
|
|
March
1, 2018
|
|
/s/Anthony J.
Cataldo
Anthony
J. Cataldo
|
|
Director
|
|
March
1, 2018
|
|
/s/ Geoffrey
Davis
Geoffrey
Davis
|
|
Director
|
|
March
1, 2018
|
GT BIOPHARMA, INC. AND SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS
YEARS ENDED DECEMBER 31, 2017 AND 2016
Contents
|
|
Page
|
|
Report
of Independent Registered Public Accounting Firm
|
|
|
Seligson
& Giannattasio, LLP
|
F-1
|
|
Consolidated
Financial Statements
|
|
|
Balance
Sheets as of December 31, 2017 and 2016
|
F-2
|
|
Statements
of Operations For Years Ended December 31, 2017 and
2016
|
F-3
|
|
Statement
of Stockholders’ Equity (Deficit) For Years Ended December
31, 2017 and 2016
|
F-4
|
|
Statements
of Cash Flows For Years Ended December 31, 2017 and
2016
|
F-5
|
|
Notes
To Consolidated Financial Statements
|
F-6
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING
FIRM
To
the shareholders and the board of directors of
GT Biopharma, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of GT
Biopharma, Inc. (the "Company") and subsidiaries as of
December 31, 2017 and 2016, the related consolidated statements of
operations, stockholders’ deficit and cash flows for each of
the two years in the period ended December 31, 2017, and the
related notes (collectively referred to as the “financial
statements”. In our opinion, the consolidated financial
statements present fairly, in all material respects, the
consolidated financial position of the Company and subsidiaries as
of December 31, 2017 and 2016 and the consolidated results of
its operations and its consolidated cash flows for each of the two
years in the period ended December 31, 2017, in conformity with
accounting principles generally accepted in the United States of
America.
Basis of Opinion
These financial statements are the responsibility of the
Company’s management. Our responsibility is to express an
opinion on the Company’s consolidated financial statements
based on our audits. We are a public accounting firm registered
with the Public Company Accounting Oversight Board (United States)
(“PCAOB”) and are required to be independent with
respect to the Company in accordance with the U.S. federal
securities laws and the applicable rules and regulations of the
Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the
PCAOB. Those standards require that we plan and perform the audit
to obtain reasonable assurance about whether the consolidated
financial statements are free of material misstatement, whether due
to error or fraud. The Company is not required to have, nor were we
engaged to perform, an audit of its internal control over financial
reporting. As part of our audits we are required to obtain an
understanding of internal control over financial reporting but not
for the purpose of expressing an opinion on the effectiveness of
the Company’s internal control over financial reporting.
Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of
material misstatement of the financial statements, whether due to
error or fraud, and performing procedures that respond to those
risks. Such procedures included examining, on a test basis,
evidence regarding the amounts and disclosures in the financial
statements. Our audits also included evaluating the accounting
principles used and significant estimates made by management, as
well as evaluating the overall presentation of the financial
statements. We believe that our audits provide a reasonable basis
for our opinion.
The accompanying consolidated financial statements have been
prepared assuming the Company will continue as a going concern. As
discussed in Note 1 to the financial statements, the Company has
incurred significant recurring losses. The realization of a major
portion of its assets is dependent upon its ability to meet its
future financing needs and the success of its future operations.
These factors raise substantial doubt about the Company’s
ability to continue as a going concern. The consolidated financial
statements do not include any adjustments that might result from
this uncertainty.
/s/ Seligson & Giannattasio, LLP
Seligson & Giannattasio, LLP
We have served as the Company’s auditor since
2008.
White Plains, New York
March 1, 2018
GT Biopharma, Inc. and Subsidiaries
December 31, 2017 and 2016
Consolidated Balance Sheets
|
|
|
|
|
ASSETS
|
|
|
|
Current
Assets:
|
|
|
|
Cash
and cash equivalents
|
$576,000
|
$19,000
|
|
Prepaid
expenses
|
-
|
2,000
|
|
Total
Current Assets
|
576,000
|
21,000
|
|
|
|
|
|
Intangible
assets
|
253,777,000
|
-
|
|
Deposits
|
9,000
|
-
|
|
Fixed
assets, net
|
6,000
|
4,000
|
|
Total
Other Assets
|
253,792,000
|
4,000
|
|
TOTAL
ASSETS
|
$254,368,000
|
$25,000
|
|
LIABILITIES AND STOCKHOLDERS’ DEFICIT
|
|
|
|
Current
Liabilities:
|
|
|
|
Accounts
payable
|
$2,546,000
|
$2,100,000
|
|
Accrued
interest
|
-
|
3,800,000
|
|
Accrued
expenses
|
102,000
|
219,000
|
|
Line
of credit
|
31,000
|
31,000
|
|
Warrant
liability
|
-
|
417,000
|
|
Settlement
note payable
|
-
|
691,000
|
|
Demand
notes payable, net of discount of $-0- and $-0-
|
-
|
452,000
|
|
Convertible
debentures, net of discount of $-0- and $794,000 current
portion
|
-
|
10,350,000
|
|
Senior
secured convertible debentures
|
-
|
889,000
|
|
Total
Current Liabilities
|
2,679,000
|
18,949,000
|
|
|
|
|
|
Long
term liabilities:
|
|
|
|
Convertible debentures, net of discount of $-0- and
$2,536,000
|
-
|
-
|
|
Total
long term liabilities
|
-
|
0
|
|
Total
liabilities
|
2,679,000
|
18,949,000
|
|
|
|
|
|
Stockholders’
Deficit:
|
|
|
|
Convertible
preferred stock - $0.001 par value; 15,000,000 shares
authorized:
|
|
|
|
Series
C - 96,230 and 96,230 shares issued and outstanding at December 31,
2017 and December 31, 2016, respectively
|
1,000
|
1,000
|
|
Series
H – 25,000 shares issued and outstanding at December 31,
2016
|
-
|
-
|
|
Series
I – 1,666,667 shares issued and outstanding December 31,
2016
|
-
|
2,000
|
|
Series
J – 1,163,548 shares issued and outstanding at December 31,
2017
|
1,000
|
-
|
|
Common
stock - $0.001 par value; 750,000,000 shares authorized; and
50,117,977 and 104,218 shares issued and outstanding at December
31, 2017 and December 31, 2016, respectively
|
50,000
|
0
|
|
Additional
paid-in capital
|
519,702,000
|
105,891,000
|
|
Accumulated
deficit
|
(267,896,000)
|
(124,649,000)
|
|
Noncontrolling
interest
|
(169,000)
|
(169,000)
|
|
Total
Stockholders’ Deficit
|
251,689,000
|
(18,924,000)
|
|
TOTAL
LIABILITIES AND STOCKHOLDERS’ DEFICIT
|
$254,368,000
|
$25,000
|
The accompanying notes are an integral part of these consolidated
financial statements.
GT Biopharma, Inc. and Subsidiaries
December 31, 2017 and 2016
Statements of Operations
|
|
|
|
|
|
|
|
Revenue:
|
|
|
|
Product
revenues
|
$-
|
$-
|
|
License
revenues
|
-
|
-
|
|
TOTAL
REVENUE
|
-
|
-
|
|
Cost
of Product Revenue
|
-
|
-
|
|
Gross
profit
|
-
|
-
|
|
Operating
Expenses:
|
|
|
|
Research
and development
|
1,068,000
|
975,000
|
|
Selling,
general and administrative
|
134,502,000
|
8,399,000
|
|
Total
operating expenses
|
135,570,000
|
9,374,000
|
|
Loss
from Operations
|
( 135,570,000)
|
( 9,374,000)
|
|
Other
income (expense)
|
|
|
|
Change
in value of warrant and derivative liabilities
|
925,000
|
25,697,000
|
|
Interest
expense/income
|
( 8,602,000)
|
( 6,555,000)
|
|
Total
Other Income (Expense)
|
( 7,677,000)
|
19,142,000
|
|
Income
(loss) before minority interest and provision for
|
|
|
|
income taxes
|
( 143,247,000)
|
9,768,000
|
|
Less:
Net loss attributable to the noncontrolling interests
|
-
|
-
|
|
Income
(loss) before provision for income taxes
|
( 143,247,000)
|
9,768,000
|
|
Provision
for income taxes
|
-
|
-
|
|
Net
income (loss)
|
( 143,247,000)
|
9,768,000
|
|
|
|
|
|
Weighted
average shares outstanding
|
|
|
|
Basic
|
16,769,431
|
81,460
|
|
Diluted
|
16,769,431
|
81,460
|
|
|
|
|
|
Net
income (loss) per share
|
|
|
|
Basic
|
$(8.54)
|
$119.91
|
|
Diluted
|
$(8.54)
|
$119.91
|
The accompanying notes are an integral part of these consolidated
financial statements.
|
GT Biopharma, Inc. and Subsidiaries
|
|
Consolidated Statement of Stockholders’ Deficit
|
|
For the Years Ended December 31, 2017 and 2016
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance
at December 31, 2015
|
1,787,897
|
$3,000
|
8,000
|
$-0
|
$84,014,000
|
$(134,417,000)
|
|
Issuance of stock
options
|
|
|
|
|
42,000
|
|
|
common stock for
convertible notes and interest
|
|
|
18,012
|
-
|
2,489,000
|
|
|
Issuance of common
stock for warrants
|
|
|
52,091
|
-
|
9,043,000
|
|
|
Issuance of common
stock for compensation
|
|
|
26,115
|
-
|
10,303,000
|
|
|
Net
income
|
|
|
|
|
|
9,768,000
|
|
Balance
at December 31, 2016
|
1,787,897
|
$3,000
|
104,218
|
$-
|
$105,891,000
|
$(124,649,000)
|
|
Issuance of common
stock for acquisition
|
|
|
16,927,878
|
17,000
|
253,901,000
|
|
|
Issuance of common
and preferred stock for convertible notes and interest
|
908,502
|
1,000
|
17,677,904
|
18,000
|
24,329,000
|
|
|
Issuance of common
and preferred stock for warrants
|
5,046
|
|
496,855
|
-
|
5,819,000
|
|
|
Issuance of common
stock for preferred stock
|
(2,041,667)
|
(2,000)
|
5,677,734
|
6,000
|
(4,000)
|
|
|
Issuance of common
and preferred stock for compensation
|
600,000
|
|
9,233,388
|
9,000
|
129,766,000
|
(143,247,000)
|
|
Net
loss
|
|
|
|
|
|
|
|
Balance
at December 31, 2017
|
1,259,778
|
$2,000
|
50,117,977
|
$50,000
|
$519,702,000
|
$(267,896,000)
|
The accompanying notes are an integral part of these consolidated
financial statements.
GT Biopharma, Inc. and Subsidiaries
Consolidated Statements of Cash Flows
For the Years Ended December 31, 2017 and 2016
|
|
|
|
|
CASH
FLOWS FROM OPERATING ACTIVITIES:
|
|
|
|
Net
income (loss)
|
$(143,247,000)
|
$9,768,000
|
|
Adjustments
to reconcile net income (loss) to net cash used in operating
activities:
|
|
|
|
Depreciation
|
2,000
|
1,000
|
|
Amortization
of intangible assets
|
-
|
-
|
|
Stock
compensation expense for options and warrants issued to
employees and non-employees
|
130,124,000
|
6,591,000
|
|
Note
Allonges
|
100,000
|
65,000
|
|
Amortization
of debt discounts
|
4,914,000
|
2,897,000
|
|
Non-cash
interest expense
|
2,197,000
|
1,632,000
|
|
Change
in value of warrant and derivative liabilities
|
( 925,000)
|
(25,697,000)
|
|
Note
settlement
|
-
|
-
|
|
Changes
in operating assets and liabilities:
|
|
|
|
Inventory
|
-
|
-
|
|
Other
assets
|
(7,000)
|
-
|
|
Accounts
payable and accrued liabilities
|
1,412,000
|
2,813,000
|
|
Net
cash used in operating activities
|
(5,430,000)
|
(1,930,000)
|
|
CASH
FLOWS FROM INVESTING ACTIVITIES:
|
|
|
|
Acquisition
of fixed assets
|
(4,000)
|
-
|
|
Net
cash used by investing activities
|
(4,000)
|
-
|
|
CASH
FLOWS FROM FINANCING ACTIVITIES:
|
|
|
|
Proceeds
from notes payable
|
5,991,000
|
1,902,000
|
|
Repayment
of note payable
|
-
|
-
|
|
Net
cash provided by financing activities
|
5,991,000
|
1,902,000
|
|
Minority
interest
|
-
|
-
|
|
NET
INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS
|
557,000
|
(28,000)
|
|
CASH
AND CASH EQUIVALENTS - Beginning of period
|
19,000
|
47,000
|
|
CASH
AND CASH EQUIVALENTS - End of period
|
$576,000
|
$19,000
|
|
|
|
|
|
Supplemental
disclosures:
|
|
|
|
Issuance
of common stock for interest expense
|
$5,179,000
|
$528,000
|
|
Issuance
of common stock for debt
|
$19,166,000
|
$1,944,000
|
The accompanying condensed notes are an integral part of these
consolidated financial statements.
1.
The Company and Summary of Significant Accounting
Policies
We are an immuno-oncology
company with a close-to-market central nervous system, or CNS,
portfolio of products. Our immuno-oncology portfolio is based off a
robust technology platform consisting of single-chain bi-, tri- and
tetra-specific scFv’s, combined with proprietary
antibody-drug linkers and drug payloads. Constructs include
bispecific and trispecific scFv constructs, , proprietary drug
payloads, bispecific targeted antibody-drug conjugates, or ADCs, as
well as tri- and tetra-specific antibody-directed cellular
cytotoxicity, or ADCC. Our proprietary tri- and tetra-specific ADCC
platform engages natural killer cells, or NK cells. NK cells are
cytotoxic lymphocytes of the innate immune system capable of immune
surveillance. NK cells mediate ADCC through the highly potent CD16
activating receptor. Upon activation, NK cells deliver a store of
membrane penetrating apoptosis-inducing molecules. Unlike T cells,
NK cells do not require antigen priming.
Our CNS portfolio consists of innovative reformulations and/or
repurposing of existing therapies. These new therapeutic agents
address numerous unmet medical needs that can lead to improved
efficacy while addressing tolerability and safety issues that
tended to limit the usefulness of the original approved drug. These
CNS drug candidates address disease states such as chronic
neuropathic pain, myasthenia gravis and vestibular
disorders.
In 1965, the corporate predecessor of GT Biopharma, Diagnostic
Data, Inc. was incorporated in the State of California. Diagnostic
Data changed its incorporation to the State of Delaware in 1972;
and changed its name to DDI Pharmaceuticals, Inc. in 1985. In 1994,
DDI Pharmaceuticals merged with International BioClinical, Inc. and
Bioxytech S.A. and changed its name to OXIS International, Inc. In
July 2017, the Company changed its name to GT Biopharma,
Inc.
Going Concern
As
shown in the accompanying consolidated financial statements, the
Company has incurred an accumulated deficit of $267,896,000 through
December 31,
2017. On a consolidated basis, the Company had
cash and cash equivalents of $576,000 at December 31, 2017. The Company's plan is to raise additional capital
until such time that the Company generates sufficient revenues to
cover its cash flow needs and/or it achieves profitability.
However, the Company cannot assure that it will accomplish this
task and there are many factors that may prevent the Company from
reaching its goal of profitability.
The current rate of cash usage raises substantial doubt about the
Company’s ability to continue as a going concern, absent any
sources of significant cash flows. In an effort to mitigate this
near-term concern the Company intends to seek additional equity or
debt financing to obtain sufficient funds to sustain operations.
However, the Company cannot provide assurance that it will
successfully obtain equity or debt or other financing, if any,
sufficient to finance its goals or that the Company will generate
future product related revenues. The Company’s financial
statements do not include any adjustments relating to the
recoverability and classification of recorded assets, or the
amounts and classification of liabilities that might be necessary
in the event that the Company cannot continue in
existence.
Advertising and promotional fees
Advertising expenses consist primarily of costs incurred in the
design, development, and printing of Company literature and
marketing materials. The Company expenses all advertising
expenditures as incurred. There were no advertising expenses for
the years ended December 31, 2017 and 2016,
respectively.
Basis of Consolidation and Comprehensive Income
The accompanying consolidated financial statements include the
accounts of GT Biopharma, Inc. and its subsidiaries. All
intercompany balances and transactions have been eliminated. The
Company's financial statements are prepared using the accrual
method of accounting.
Cash and Cash Equivalents
The Company considers all highly liquid investments with original
maturities of three months or less to be cash
equivalents.
Concentrations of Credit Risk
The Company's cash and cash equivalents, marketable securities and
accounts receivable are monitored for exposure to concentrations of
credit risk. The Company maintains substantially all of its cash
balances in a limited number of financial institutions. The
balances are each insured by the Federal Deposit Insurance
Corporation up to $250,000. The Company had $316,000 of balances in
excess of this limit at December 31, 2017.
Fair Value of Financial Instruments
The carrying amounts of cash and cash equivalents, restricted cash,
accounts receivable, inventory, accounts payable and accrued
expenses approximate fair value because of the short-term nature of
these instruments. The fair value of debt is based upon current
interest rates for debt instruments with comparable maturities and
characteristics and approximates the carrying amount.
Stock Based Compensation to Employees
The
Company accounts for its stock-based compensation for employees in
accordance with Accounting Standards Codification
(“ASC”) 718. The Company recognizes in the
statement of operations the grant-date fair value of stock options
and other equity-based compensation issued to employees and
non-employees over the related vesting period.
Impairment of Long Lived Assets
The Company's long-lived assets currently consist of capitalized
patents. The Company evaluates its long-lived assets for impairment
whenever events or changes in circumstances indicate that the
carrying amount of such assets may not be recoverable. If any of
the Company's long-lived assets are considered to be impaired, the
amount of impairment to be recognized is equal to the excess of the
carrying amount of the assets over the fair value of the
assets.
Income Taxes
The Company accounts for income taxes using the asset and liability
approach, whereby deferred income tax assets and liabilities are
recognized for the estimated future tax effects, based on current
enacted tax laws, of temporary differences between financial and
tax reporting for current and prior periods. Deferred tax assets
are reduced, if necessary, by a valuation allowance if the
corresponding future tax benefits may not be realized.
Net Income (Loss) per Share
Basic net income (loss) per share is computed by dividing the net
loss for the period by the weighted average number of common shares
outstanding during the period. Diluted net income (loss) per share
is computed by dividing the net loss for the period by the weighted
average number of common shares outstanding during the period, plus
the potential dilutive effect of common shares issuable upon
exercise or conversion of outstanding stock options and warrants
during the period. The weighted average number of potentially
dilutive common shares excluded from the calculation of net income
(loss) per share totaled in 1,164,795 in 2017 and 126,145 in
2016.
Goodwill and Other Intangible Assets
Certain intangible assets were acquired as part of a business
combination (see note 3) and have been capitalized at their
acquisition date fair value of $253,777,000, based on the share
value of the Company’s stock issued for the acquisition of
the assets. Acquired definite life intangible assets are amortized
using the straight-line method over their respective estimated
useful lives. The Company evaluates the potential
impairment of intangible assets if events or changes in
circumstances indicate that the carrying amount of the assets may
not be fully recoverable or that the useful lives of these assets
are no longer appropriate. Goodwill is not amortized but is
evaluated for impairment within the Company’s single
reporting unit on an annual basis, during the fourth quarter, or
more frequently if an event occurs or circumstances change that
would more-likely-than-not reduce the fair value of the
Company’s reporting unit below its carrying
amount.
Patents
Acquired patents are capitalized at their acquisition cost or fair
value. The legal costs, patent registration fees and models and
drawings required for filing patent applications are capitalized if
they relate to commercially viable technologies. Commercially
viable technologies are those technologies that are projected to
generate future positive cash flows in the near term. Legal costs
associated with patent applications that are not determined to be
commercially viable are expensed as incurred. All research and
development costs incurred in developing the patentable idea are
expensed as incurred. Legal fees from the costs incurred in
successful defense to the extent of an evident increase in the
value of the patents are capitalized.
Capitalized cost for pending patents are amortized on a
straight-line basis over the remaining twenty year legal life of
each patent after the costs have been incurred. Once each patent is
issued, capitalized costs are amortized on a straight-line basis
over the shorter of the patent's remaining statutory life,
estimated economic life or ten years.
Fixed Assets
Fixed assets are stated at cost. Depreciation is computed on a
straight-line basis over the estimated useful lives of the assets,
which are 3 to 10 years for machinery and equipment and the
shorter of the lease term or estimated economic life for leasehold
improvements.
Fair Value
The carrying amounts reported in the balance sheets for receivables
and current liabilities each qualify as financial instruments and
are a reasonable estimate of fair value because of the short period
of time between the origination of such instruments and their
expected realization and their current market rate of
interest. The three levels are defined as
follows:
●
Level 1 inputs to
the valuation methodology are quoted prices (unadjusted) for
identical assets or liabilities in active markets. The
Company’s Level 1 assets include cash equivalents, primarily
institutional money market funds, whose carrying value represents
fair value because of their short-term maturities of the
investments held by these funds.
●
Level 2 inputs to
the valuation methodology include quoted prices for similar assets
and liabilities in active markets, and inputs that are observable
for the asset or liability, either directly or indirectly, for
substantially the full term of the financial instrument. The
Company’s Level 2 liabilities consist of liabilities arising
from the issuance of convertible securities and in accordance with
ASC 815-40: a warrant liability for detachable warrants, as well as
an accrued derivative liability for the beneficial conversion
feature. These liabilities are remeasured each reporting period.
Fair value is determined using the Binomial valuation model based
on observable market inputs, such as share price data and a
discount rate consistent with that of a government-issued security
of a similar maturity. There were not such liabilities at December
31, 2017.
●
Level 3 inputs to
the valuation methodology are unobservable and significant to the
fair value measurement.
Research and Development
Research and development costs are expensed as incurred and
reported as research and development expense. Research and
development costs totaling $1,068,000 and $975,000 for the years
ended December 31, 2017 and 2016, respectively.
Revenue Recognition
License Revenue
License
arrangements may consist of non-refundable upfront license fees,
exclusive licensed rights to patented or patent pending technology,
and various performance or sales milestones and future product
royalty payments. Some of these arrangements are multiple element
arrangements.
Non-refundable,
up-front fees that are not contingent on any future performance by
us, and require no consequential continuing involvement on our
part, are recognized as revenue when the license term commences and
the licensed data, technology and/or compound is
delivered. We defer recognition of non-refundable
upfront fees if we have continuing performance obligations without
which the technology, right, product or service conveyed in
conjunction with the non-refundable fee has no utility to the
licensee that is separate and independent of our performance under
the other elements of the arrangement. In addition, if we have
continuing involvement through research and development services
that are required because our know-how and expertise related to the
technology is proprietary to us, or can only be performed by us,
then such up-front fees are deferred and recognized over the period
of continuing involvement.
Payments
related to substantive, performance-based milestones in a research
and development arrangement are recognized as revenue upon the
achievement of the milestones as specified in the underlying
agreements when they represent the culmination of the earnings
process.
Use of Estimates
The
financial statements and notes are representations of the Company's
management, which is responsible for their integrity and
objectivity. These accounting policies conform to accounting
principles generally accepted in the United States of America and
have been consistently applied in the preparation of the financial
statements. The preparation of financial statements requires
management to make estimates and assumptions that affect the
reported amounts of assets, liabilities revenues and expenses and
disclosures of contingent assets and liabilities at the date of the
financial statements. Actual results could differ from those
estimates.
Recently Issued Standards
In
February 2016, the Financial Accounting Standards Board
(“FASB”) issued Accounting Standard Update
(“ASU”) No. 2016-02, “Leases.” This ASU
requires all lessees to be recognized on the balance sheet as right
to use assets and lease liabilities for the rights and obligations
created by lease arrangements with terms greater than 12 months.
The amendments in this ASU are effective for fiscal years beginning
after December 15, 2018 and for interim periods therein. The
Company is in the process of assessing the impact the adoption this
ASU will have on its consolidated financial position, results of
operations and cash flows. At a minimum, total assets and total
liabilities will increase in the period the ASU is adopted. Early
adoption of this ASU is permitted. At December 31, 2017, the
Company’s undiscounted future minimum payments outstanding
for lease obligations (including those currently included as
capital lease obligations) were
approximately $297,000.
In May
2014, the FASB issued ASU No. 2014-09, “Revenue from
Contracts with Customers: Topic 606.” This ASU replaces
nearly all existing U.S. GAAP guidance on revenue recognition. The
standard prescribes a five-step model for recognizing revenue, the
application of which will require significant judgment. The
amendments in this ASU are effective for fiscal years beginning
after December 15, 2017, and for interim periods therein. The
provisions of this ASU may be applied retroactively or on a
modified retrospective (cumulative effect) basis. The Company will
adopt the standard using the modified retrospective approach in its
fiscal year beginning January 1, 2018. Adoption of this ASU will
not have a significant impact on the Company’s consolidated
financial position, results of operations and cash
flows.
Senior secured convertible debentures
On
October 25, 2006, the Company entered into a securities purchase
agreement (“2006 Purchase Agreement”) with four
accredited investors (the “2006 Purchasers”). In
conjunction with the signing of the 2006 Purchase Agreement, the
Company issued secured convertible debentures (“2006
Debentures”) and Series A, B, C, D, and E common stock
warrants (“2006 Warrants”) to the 2006 Purchasers, and
the parties also entered into a security agreement (the “2006
Security Agreement”) pursuant to which the Company agreed to
grant the 2006 Purchasers, pari passu, a security interest in
substantially all of the Company’s assets.
Pursuant
to the terms of the 2006 Purchase Agreement, the Company issued the
2006 Debentures in an aggregate principal amount of $1,694,250 to
the 2006 Purchasers. The 2006 Debentures are subject to an original
issue discount of 20.318% resulting in proceeds to the Company of
$1,350,000 from the transaction. The 2006 Debentures were due on
October 25, 2008. The 2006 Debentures are convertible, at the
option of the 2006 Purchasers, at any time prior to payment in
full, into shares of common stock of the Company. As a result of
the full ratchet anti-dilution provision the current conversion
price is the lesser of $0.40 or 60% of the average of the lowest
three trading prices occurring at any time during the 20 trading
days preceding conversion (the “2006 Conversion
Price”). Beginning on the first of the month beginning
February 1, 2007, the Company was required to amortize the 2006
Debentures in equal installments on a monthly basis resulting in a
complete repayment by the maturity date (the “Monthly
Redemption Amounts”). The Monthly Redemption Amounts could
have been paid in cash or in shares, subject to certain
restrictions. If the Company chose to make any Monthly Redemption
Amount payment in shares of common stock, the price per share would
have been the lesser of the Conversion Price then in effect and 85%
of the weighted average price for the 10-trading days prior to the
due date of the Monthly Redemption Amount. The Company did not make
any of the required monthly redemption payments.
Pursuant
to the provisions of the 2006 Debentures, such non-payment was an
event of default and penalty interest has accrued on the unpaid
redemption balance at an interest rate equal to the lower of 18%
per annum and the maximum rate permitted by applicable law. In
addition, each of the 2006 Purchasers has the right to accelerate
the cash repayment of at least 130% of the outstanding principal
amount of the 2006 Debenture (plus accrued but unpaid liquidated
damages and interest) and to sell substantially all of the
Company’s assets pursuant to the provisions of the 2006
Security Agreement to satisfy any such unpaid balance.
The
Company and Bristol entered into a Forbearance Agreement on
December 3, 2015, pursuant to which Bristol agreed to refrain and
forbear from exercising certain rights and remedies with respect
the 2006 Debentures for three months. In exchange for the
Forbearance Agreement, the Company issued an allonge in the amount
of $350,000 increasing the principal amount if the 2006
Debentures.
During the year ended December 31, 2017 the Company converted the
remaining balance of $889,000 of the 2006 Debentures into common
stock of the Company.
Convertible debentures
From
October 2009 to September 2016, the Company has entered into
multiple convertible debenture arrangements with several accredited
investors (“Convertible Debentures”). Interest on the
Convertible Debentures ranges for 0% to 18% with a default rate of
18%. The Convertible Debentures are either two year or six month
notes.
The
conversion price of the Convertible Debentures is subject to full
ratchet anti-dilution adjustment in the event that the Company
thereafter issues common stock or common stock equivalents at a
price per share less than the conversion price or the exercise
price, respectively, and to other normal and customary
anti-dilution adjustment upon certain other events. As a result of
the full ratchet anti-dilution provision the current conversion
price is $0.40 per share and the default conversion price is 65% of
the average of the lowest three trading prices occurring at any
time during the 20 trading days preceding conversion .
The
holders of the Convertible Debentures have contractually agreed to
restrict their ability to convert their Convertible Debentures and
receive shares of our common stock such that the number of shares
of the Company common stock held by holders and its affiliates
after such conversion or exercise does not exceed 4.9% or 9.9% of
the Company’s then issued and outstanding shares of common
stock. On
August 31, 2017, the Company converted all Convertible Debentures
into common and Series J preferred stock of the
Company.
Convertible
Debentures held by the Company are as follows:
|
|
Balance
at
December
31,
2017
|
Balance
at
December
31,
2016
|
|
|
|
|
|
2009
Debentures
|
$-
|
$305,000
|
|
June 2011
Debentures
|
-
|
64,000
|
|
November 2011
Debentures
|
-
|
125,000
|
|
March 2012
Debentures
|
-
|
140,000
|
|
May 2012
Debentures
|
-
|
225,000
|
|
December 2012
Debentures
|
-
|
425,000
|
|
November 2013
Debentures
|
-
|
172,000
|
|
July 2014
Debentures
|
-
|
3,140,000
|
|
October 2014
Debentures
|
-
|
1,250,000
|
|
March 2015
Debentures
|
-
|
2,175,000
|
|
July 2015
Debentures
|
-
|
500,000
|
|
October 2015
Debentures
|
-
|
330,000
|
|
November 2015
Debentures
|
-
|
190,000
|
|
December 2015
Debentures
|
-
|
200,000
|
|
January 2016
Debentures
|
-
|
150,000
|
|
May 2016
Debentures
|
-
|
1,503,000
|
|
September 2016
Debentures
|
-
|
250,000
|
|
January 2017
Debentures
|
-
|
-
|
|
March 2017
Debentures
|
-
|
-
|
|
April 2017
Debentures
|
-
|
-
|
|
July 2017
Debentures
|
-
|
-
|
|
August 2017
Debentures
|
-
|
|
|
|
|
|
|
Total convertible
debentures
|
$-
|
$11,144,000
|
|
Less:
discount
|
-
|
(794,000)
|
|
Total convertible
debentures, net of discount
|
$-
|
$10,350,000
|
|
|
|
|
|
Total short term
convertible debentures, net of discount
|
$-
|
$10,350,000
|
Settlement Note Payable
On
August 8, 2012, a Settlement Agreement and Mutual General Release
("Agreement") was made by and between OXIS and Bristol Investment
Fund, Ltd., in order to settle certain claims regarding certain
convertible debentures held by Bristol.
Pursuant
to the Agreement, OXIS shall pay Bristol (half of which payment
would redound to Theorem Capital LLC (“Theorem”)) a
total of $1,119,778 as payment in full for the losses suffered and
all costs incurred by Bristol in connection with the Transaction.
Payment of such $1,119,778 shall be made as follows: OXIS shall
issue restricted common stock to each of Bristol and Theorem, in an
amount such that each Bristol and Theorem shall hold no more than
9.99% of the outstanding shares of OXIS (including any shares that
each may hold as of the date of issuance). The shares so issued
represent $417,475.65 of the $1,119,778 payment (111,327 shares at
$3.75 per share, of which 36,675 will be retained by Bristol and
74,652 will be issued to Theorem). The remaining balance of the
payment shall be made in the form of two convertible promissory
notes in the respective amounts of $422,357.75 for Bristol and
$279,944.60 for Theorem (collectively, the “Notes”)
with a maturity of December 1, 2017 having an 8% annual interest
rate, with interest only accruing until January 1, 2013, and then
level payments of $3,750 each beginning January 1, 2013 until paid
in full on December 1, 2017. In the event a default in the monthly
payments on the Notes has occurred and is continuing each holder of
the Notes shall be permitted to convert the unpaid principal and
interest of the Notes into shares of OXIS at $0.40 cents per share.
In the absence of such continuing default no conversion of the
Notes will be permitted. OXIS will have the right to repay the
Notes in full at any time without penalty. On August 31,2017 the
Company converted the remaining balance of $691,000 of the
Settlement Note Payable into common stock of the
Company.
Demand Notes
On
February 7, 2011 the Company entered into a convertible demand
promissory note with Bristol pursuant to which Bristol purchased an
aggregate principal amount of $31,375 of convertible demand
promissory notes for an aggregate purchase price of $25,000 (the
“February 2011 Bristol Note”). The February 2011
Bristol Note is convertible into shares of common stock of the
Company at a price equal to the lesser of (i) $15.00 or (ii) the
average of the three (3) lowest intra-day trading prices of the
Common Stock during the 20 Trading Days immediately prior to the
date on which the Notice of Conversion is delivered to the Company.
During the
quarter ended March 31, 2017 the Company converted the entire
balance of $31,375 into common stock of the
Company.
On
March 4, 2011 the Company entered into a convertible demand
promissory note with Bristol pursuant to which Bristol purchased an
aggregate principal amount of $31,375 of convertible demand
promissory notes for an aggregate purchase price of $25,000 (the
“March 2011 Bristol Note”). The March 2011 Bristol Note
is convertible at the option of the holder at any time into shares
of common stock, at a price equal to the lesser of (i) $15.00 or
(ii) the average of the three (3) lowest intra-day trading prices
of the Common Stock during the 20 Trading Days immediately prior to
the date on which the Notice of Conversion is delivered to the
Company. During the quarter
ended March 31, 2017 the Company converted the entire balance of
$31,375 into common stock of the Company.
On
October 26, 2011 the Company entered into a convertible demand
promissory note with Theorem pursuant to which Theorem purchased an
aggregate principal amount of $200,000 of convertible demand
promissory notes for an aggregate purchase price of $157,217 (the
“October 2011 Theorem Note”). The October 2011 Theorem
Note is convertible into shares of common stock of the Company, at
a price equal to the lesser of (i) $15.00 or (ii) the average of
the three (3) lowest intra-day trading prices of the Common Stock
during the 20 Trading Days immediately prior to the date on which
the Notice of Conversion is delivered to the Company. During the quarter
ended March 31, 2017 the Company converted the entire balance of
$200,000 into common stock of the
Company.
In
December, 2013, the Company entered into a convertible demand
promissory note with an initial principal balance of $189,662
convertible at a price equal to the lesser of (i) $15.00 or (ii)
the average of the three (3) lowest intra-day trading prices of the
Common Stock during the 20 Trading Days immediately prior to the
date on which the Notice of Conversion is delivered to the Company.
On August
31,2017, the Company converted the remaining balance of $189,662 of
this Demand Note Payable into common stock of the
Company.
Financing
Agreement
On
November 8, 2010, the Company entered into a financing arrangement
with Gemini Pharmaceuticals, Inc., a product development and
manufacturing partner of the Company, pursuant to which Gemini
Pharmaceuticals made a $250,000 strategic equity investment in the
Company and agreed to make a $750,000 purchase order line of credit
facility available to the Company. The outstanding principal of all
Advances under the Line of Credit will bear interest at the rate of
interest of prime plus 2 percent per annum. There is $31,000 due on
this credit line at December 31, 2017.
Stock Split
In July 2017, the Company approved a one for three hundred reverse
stock split. The Company has reported the effect of the split
retroactively for all periods presented.
Common Shares
In July 2017, the Company amended its articles of incorporation to
change the number of authorized common shares to 750,000,000 shares
of $.001 par value stock.
Common Stock
During the year ended December 31, 2016, the Company issued
an aggregate of 41,934 shares
of common stock to a total of 34 persons or entities in exchange of
the cancellation of warrants on a cashless
basis.
During the year ended December 31, 2016, the Company also issued
an aggregate of 6,741 shares of
common stock to a total of 17 persons as payment for consulting
services provided to the Company. The average valuation
of these shares was $600.00 per share.
During the year ended December 31, 2016, the Company also issued an
aggregate of 15,375 shares of common stock to two executive
officers of the Company in fulfilment of contractual rights held by
the officers pursuant to their employment
agreements.
During the year ended December 31, 2016, the Company also issued
an aggregate of 19,857 shares
of common stock to a total of 18 persons as payment for the
conversion of certain note and the related accrued
interest. The conversion price of these shares was
$120.00 per share.
In August 2016, the Company issued 3,717 shares of common stock to H.C. Wainwright and
Co., LLC as payment for investment banking services provided to the
Company.
In October 2016, the Company issued an aggregate of 1,982 shares of common stock to
one noteholder as payment for the conversion of a certain
note. The conversion price of these shares was $25.23
per share based on 60% of the average of the lowest three
trading prices occurring at any time during the 20 trading days
preceding conversion.
In November 2016, the Company issued an aggregate of 3,250 shares of common stock to
one noteholder as payment for the conversion of a certain
note. The conversion price of these shares was $15.39
per share based on 60% of the average of the lowest three
trading prices occurring at any time during the 20 trading days
preceding conversion.
In December 2016, the Company issued an aggregate of 3,414 shares of common stock to
one noteholder as payment for the conversion of a certain
note. The conversion price of these shares was $14.65
per share based on 60% of the average of the lowest three
trading prices occurring at any time during the 20 trading days
preceding conversion.
All shares issued during 2016 were exempt from the registration
requirements of Section 5 of the Securities Act of 1933 (the
“Act”) pursuant to Section 4(2) of the Act since
the shares were issued to persons or entities closely associated
with the Company and there was no public offering of the
shares.
On September 1, 2017, the Company entered into an Agreement and
Plan of Merger whereby it acquired 100% of the issued and
outstanding capital stock of Georgetown Translational
Pharmaceuticals, Inc. (GTP). GTP is a biotechnology company focused
on acquiring or discovering and patenting late-stage, de-risked,
and close-to-market improved treatments for CNS disease (Neurology
and Pain) and shepherding the products through the FDA approval
process to the NDA. In exchange for the ownership of GTP, the
Company issued a total of 16,927,878 shares of its common stock to
the three prior owners of GTP which represents 33% of the issued
and outstanding capital stock of the Company.
During the six months ended June 30, 2017 the Registrant has issued
a total of 390,279 shares of common stock to a total of eleven
entities or individuals in exchange for the cancellation of debt in
the total amount of $2,025,000 and interest in the total amount of
$486,000.
In August 2017, the Company has issued a total of 17,287,625 shares
of common stock in exchange for the cancellation of debt in the
total amount of $17,141,000 and interest in the total amount of
$4,693,000.
In August 2017, the Company issued 496,855 shares of common stock
upon the exercise of warrants on a cashless
basis.
In August 2017, the Company converted 25,000 Series H and 1,666,667
Series I shares of preferred stock into 5,327,734 shares of common
stock.
In December 2017, the Company converted 350,000 Series J shares of
preferred stock into 350,000 shares of common stock.
Preferred Stock
The 96,230 shares of Series C preferred stock are convertible into
111 shares of the Company's common stock at the option of the
holders at any time. The conversion ratio is based on the average
closing bid price of the common stock for the fifteen consecutive
trading days ending on the date immediately preceding the date
notice of conversion is given, but cannot be less than .20 or more
than .2889 common shares for each Series C preferred share. The
conversion ratio may be adjusted under certain circumstances such
as stock splits or stock dividends. The Company has the right to
automatically convert the Series C preferred stock into common
stock if the Company lists its shares of common stock on the Nasdaq
National Market and the average closing bid price of the Company's
common stock on the Nasdaq National Market for 15 consecutive
trading days exceeds $3,000.00. Each share of Series C preferred
stock is entitled to the number of votes equal to .26 divided by
the average closing bid price of the Company's common stock during
the fifteen consecutive trading days immediately prior to the date
such shares of Series C preferred stock were purchased. In the
event of liquidation, the holders of the Series C preferred stock
shall participate on an equal basis with the holders of the common
stock (as if the Series C preferred stock had converted into common
stock) in any distribution of any of the assets or surplus funds of
the Company. The holders of Series C preferred stock are entitled
to noncumulative dividends if and when declared by the Company's
board of directors. No dividends to Series C preferred stockholders
were issued or unpaid through December 31, 2017.
On December 4, 2008, the Company entered into and closed an
Agreement (the “Bristol Agreement”) with Bristol
Investment Fund, Ltd. pursuant to which Bristol agreed to cancel
the debt payable by the Company to Bristol in the amount of
approximately $20,000 in consideration of the Company issuing
Bristol 25,000 shares of Series G Convertible Preferred Stock,
which such shares carry a stated value equal to $1.00 per share
(the “Series G Stock”).
The Series G Stock is convertible, at any time at the option
of the holder, into common shares of the Company based on a
conversion price equal to the lesser of $2.50 or 60% of the average
of the three lowest trading prices occurring
at any time during the 20 trading days preceding the
conversion. The Series G Stock, as
amended, shall have voting rights on an as converted basis
multiplied by 100.
In the event of any liquidation or winding up of the Company, the
holders of Series G Stock will be entitled to receive, in
preference to holders of common stock, an amount equal to the
stated value plus interest of 15% per year.
The Series G Stock restricts the ability of the holder to convert
the Series G Stock and receive shares of the Company’s
common stock such that the number of shares of the Company common
stock held by Bristol and its affiliates after such conversion does
not exceed 4.9% of the Company’s then issued and outstanding
shares of common stock.
The Series G Stock was previously referred to in an 8-K filed by
the Company on December 10, 2008 in error as the “Series E
Stock”. Further, the Series G Stock initially incorrectly
provided that it voted on an as converted basis multiplied by
10. This incorrectly reflected the intent of the Company
and the holder.
On October 13, 2009 the Company was informed by Theorem Group, LLC
that it had purchased all of the outstanding Series G Preferred
Stock and Theorem gave notice to the Company that it intended to
exercise its ability to vote on all shareholder matters utilizing
the super voting privileges provided by the Series G
Stock.
Effective
February 10, 2010, the Company issued 25,000 shares of its new
Series H Convertible Preferred Stock (the “Series H
Preferred”) to Theorem Group, LLC, a California limited
liability company (the “Stockholder”), in exchange for
the 25,000 shares of Series G Stock then owned by the
Stockholder. The foregoing exchange was effected
pursuant to that certain Exchange Agreement, dated February 10,
2010, between the Company and the Stockholder (the “Exchange
Agreement”).
The
Certificate of Designation of the Series H Preferred is based on,
and substantially similar to the form and substance of the
Certificate of Designation of the Series G
Preferred. Some of the corrections, changes and
differences between the Certificate of Designation of the Series G
Preferred and the Certificate of Designation of the Series H
Preferred include the following:
●
As previously
disclosed, the holder of the Series H Preferred is entitled to vote
with the common stock, and is entitled to a number of votes equal
to (i) the number of shares of common stock it can convert into
(without any restrictions or limitations on such conversion), (ii)
multiplied by 100.
●
The holder of the
Series H Preferred cannot convert such preferred stock into shares
of common stock if the holder and its affiliates after such
conversion would own more than 9.9% of the Company’s
then issued and outstanding shares of common stock.
●
The Series G
Preferred contained a limitation that the holder of the Series G
Preferred could not convert such preferred shares into more than
19.999% of the issued and outstanding shares of common stock
without the approval of the stockholders if the rules of the
principal market on which the common stock is traded would prohibit
such a conversion. Since the rules of the
Company’s principal market did not require such a limitation,
that provision has been deleted.
In August 2017, the Company converted 25,000 Series H stock into
5,119,401 shares of common stock.
On
November 8, 2010, the Company sold 1,666,667 shares of the
Company’s Series I Preferred Stock, $.001 par value, at a
price of $0.15 per share ($250,000).
The
holder of the Series I Preferred Stock will be entitled to receive,
out of funds legally available, dividends in cash at the annual
rate of 8.0% of the Preference Amount ($0.15), when, as, and if
declared by the Board. No dividends or other distributions shall be
made with respect to any shares of junior stock until dividends in
the same amount per share on the Series I Preferred Stock shall
have been declared and paid or set apart during that fiscal year.
Dividends on the Series I Preferred Stock shall not be cumulative
and no right shall accrue to the Series I Preferred Stock by reason
of the fact that the Company may fail to declare or pay dividends
on the Series I Preferred Stock in the amount of the Dividend Rate
per share or in any amount in any previous fiscal year of the
Company, whether or not the earnings of the Company in that
previous fiscal year were sufficient to pay such dividends in whole
or in part.
Each
share of Series I Preferred Stock shall entitle the holder thereof
to such number of votes per share as shall equal the number of
shares of Common Stock (rounded to the nearest whole number) into
which such share of Series I Preferred Stock is then
convertible.
Upon
any liquidation of the Company, subject to the rights of any series
of Preferred Stock that may from time to time come into existence,
before any distribution or payment shall be made to the holders of
any Junior Stock, the holders of the shares of Series I Preferred
Stock then outstanding shall be entitled to receive and be paid out
of the assets of the Company legally available for distribution to
its stockholders liquidating distributions in cash or property at
its fair market value as determined by the Board in the amount of
$0.15 per share (as adjusted for any stock dividends, combinations
or splits with respect to such shares).
Shares
of Series I Preferred Stock may, at the option of the holder
thereof, be converted at any time or from time to time into fully
paid and non-assessable shares of Common Stock. The number of
shares of Common Stock which a holder of shares of Series I
Preferred Stock shall be entitled to receive upon conversion of
such shares shall be the product obtained by multiplying the
Conversion Rate by the number of shares of Series I Preferred Stock
being converted. Initially, the Series I Preferred Stock is
convertible into 6,667 shares of common stock.
In the
event that the per-share Market Price of the Common Stock over a
period of 20 consecutive trading days is equal to at least 130% of
the initial conversion price (130% of $0.15), all outstanding
shares of Series I Preferred Stock shall be converted automatically
into the number of shares of Common Stock into which such shares of
Series I Preferred Stock are then convertible without any further
action by the holders of such shares and whether or not the
certificates representing such shares of Series I Preferred Stock
are surrendered to the Company or its transfer agent.
In August 2017, the Company converted 1,666,667 Series I shares of
preferred stock into 208,333 shares of common stock.
On September 1, 2017, the Company authorized 2,000,000 shares of
Series J Preferred Stock. Shares of Series J Preferred Stock
will have the same voting rights as shares of common stock with
each share of Series J Preferred Stock entitled to one vote at a
meeting of the shareholders of the Corporation. Shares of Series J
Preferred Stock will not be entitled to receive any dividends,
unless and until specifically declared by our board of directors.
The holders of the Series J Preferred Stock will participate, on an
as-if-converted-to-common stock basis, in any dividends to the
holders of common stock. Each share of the Series J Preferred Stock
is convertible into one share of our common stock at any time at
the option of the holder.
On September 1, 2017 the Company issued a total of 700,278 shares
of Series J
Preferred Stock in exchange for the
cancellation of debt in the total amount of
$840,000.
On September 1, 2017 the Company issued 5,046 shares of
Series J Preferred
Stock upon the exercise of
warrants on a cashless basis.
On September 1, 2017 the Company also issued
600,000 shares
of Series J Preferred
Stock to one entity as
payment for $720,000 of consulting services provided to the
Company.
In December 2017, the Company converted 350,000 Series J shares of
preferred stock into 350,000 shares of common stock.
Common Stock Warrants
Warrant
transactions for the years ended December 31, 2016 and 2015 are as
follows:
|
|
|
Weighted
Average Exercise Price
|
|
Outstanding,
December 31, 2015:
|
41,752
|
$375.00
|
|
Granted
|
17,005
|
135.00
|
|
Forfeited
|
(1,173)
|
375.00
|
|
Exercised
|
(42,034)
|
375.00
|
|
Outstanding at
December 31, 2016:
|
15,550
|
$135.00
|
|
Granted
|
486,351
|
15.00
|
|
Forfeited
|
|
|
|
Exercised
|
(501,901)
|
15.00
|
|
Outstanding at
December 31, 2016
|
-
|
$-
|
|
|
|
|
|
Exercisable
warrants:
|
|
|
|
December 31,
2017
|
-
|
$-
|
|
December 31,
2016
|
41,752
|
$375.00
|
Stock Options
The
Company reserved 1,333 shares of its common stock at December 31,
2014 for issuance under the 2014 Stock Incentive Plan (the
“2014 Plan”). The 2014 Plan, approval by stockholders
in May 2015, permits the Company to grant stock options to acquire
shares of the Company's common stock, award stock bonuses of the
Company's common stock, and grant stock appreciation rights. At
December 31, 2017, 445 shares of common stock were available
for grant and options to purchase 888 shares of common stock are
outstanding under the 2014 Plan.
In
addition, the Company has reserved 7 shares of its common stock for
issuance outside of its stock incentive plans. At December 31,
2017, options to purchase 7 shares of common stock are outstanding
outside of its stock incentive plans.
The
following table summarizes stock option transactions for the years
ended December 31, 2017 and 2016:
|
|
|
Weighted
Average Exercise Price
|
|
Outstanding,
December 31, 2015
|
1,249
|
$1,464.00
|
|
Granted
|
-
|
-
|
|
Exercised
|
-
|
-
|
|
Expired
|
(3)
|
16,881.00
|
|
Outstanding,
December 31, 2016
|
1,246
|
$1,428.00
|
|
Granted
|
-
|
-
|
|
Exercised
|
-
|
-
|
|
Expired
|
-
|
-
|
|
Outstanding,
December 31, 2017
|
1,246
|
$1,428.00
|
|
|
|
|
|
Exercisable
Options:
|
|
|
|
December 31,
2016
|
1,246
|
$1,428.00
|
|
December 31,
2017
|
1,246
|
$1,428.00
|
The
weighted-average fair value of options granted was $1,780,000 for
2017 and 2016.
The
following table summarizes information about all outstanding and
exercisable stock options at December 31, 2017:
|
|
|
|
|
|
|
Weighted-Average
Remaining Contractual Life
|
Weighted-Average
Exercise
Price
|
|
Weighted-Average
Exercise
Price
|
|
$750.00 to $2,225.00
|
1,246
|
1.38
|
$1,428.73
|
1,246
|
$1428.73
|
Deferred Taxes
Deferred taxes reflect the net tax effects of temporary differences
between the carrying amounts of assets and liabilities for
financial reporting purposes and the amounts used for income tax
purposes, and operating losses and tax credit carryforwards. The
significant components of net deferred income tax assets for the
Company are:
|
|
|
|
|
|
|
|
Deferred
tax assets:
|
|
|
|
Federal
net operating loss carryforward
|
$15,949,000
|
$19,819,000
|
|
Other
|
-
|
1,634,000
|
|
Patent
amortization
|
(6,000)
|
(11,000)
|
|
Deferred
tax assets before valuation
|
15,943,000
|
21,422,000
|
|
Valuation
allowance
|
(15,943,000)
|
(21,422,000)
|
|
Net
deferred income tax assets
|
$—
|
$—
|
Generally accepted accounting principles requires that the tax
benefit of net operating losses, temporary differences and credit
carryforwards be recorded as an asset to the extent that management
assesses that realization is “more likely than not.”
Realization of the future tax benefits is dependent on the
Company's ability to generate sufficient taxable income within the
carryforward period. Because of the Company's history of operating
losses, management has provided a valuation allowance equal to its
net deferred tax assets. The valuation allowance decreased by
$5,499,000 during the year ended December 31, 2017, which includes
a decrease due to a change in rates resulting from the Tax Cuts and
Jobs Act totaling $6,908,000.
Tax Carryforward
At December 31, 2017, the Company had net operating loss
carryforwards of approximately $53,165,000 to reduce United States
federal taxable income in future years. These carryforwards expire
from 2018 through 2037.
The
Company is no longer subject to U.S. and state tax examinations for
years ending before the fiscal year ended December 31, 2013.
Management does not believe there will be any material changes in
our unrecognized tax positions over the next twelve
months.
The
Company's policy is to recognize interest and penalties accrued on
any unrecognized tax benefits as a component of income tax expense.
There was no accrued interest or penalties associated with any
unrecognized tax benefits, nor was any interest expense recognized
during the years ended December 31, 2017 and 2016.
6.
Commitments and Contingencies
Leases
On September 1, 2017, the Company has entered into a three-year
lease agreement for its office in Washington, D.C. In addition to
minimum rent, certain leases require payment of real estate taxes,
insurance, common area maintenance charges and other executory
costs. These executory costs are not included in the table below.
The Company recognizes rent expense under such arrangements on a
straight-line basis over the effective term of each
lease.
The following table summarizes the Company’s future minimum
lease commitments as of December 31, 2017 (in
thousands):
|
Year ending
December 31:
|
|
|
2018
|
108,000
|
|
2019
|
108,000
|
|
2020
|
81,000
|
|
Total minimum lease
payments
|
$297,000
|
Rent expense for the year ended December 31,
2017 and 2016 was $9,000 and $-0-,
respectively.
Employment Agreements
In connection with the acquisition, we entered into employment
contracts on September 1, 2017, with Mr. Cataldo as executive
chairman, Dr. Clarence-Smith as chief executive officer, Dr.
Raymond Urbanski as chief medical officer and Steven Weldon as
chief financial officer. We entered into an employment contract on
November 15, 2017, with Mr. Cross as president and chief operating
officer.
On February 14, 2018, the Company entered into the First Amendment
to the Employment Agreement with Dr. Clarence-Smith, amending the
Employment Agreement, dated September 1, 2017, between the Company
and Dr. Clarence-Smith. Under the First Amendment, Dr.
Clarence-Smith’s title has been revised to reflect her new
position and she will be paid an annual salary of $500,000, paid in
equal monthly installment. All other terms of her original
Employment Agreement remain unchanged.
On February 14, 2018, the Company entered into a Consultant
Agreement with Mr. Cataldo. The term of the Consultant Agreement
lasts until August 31, 2020 and is terminable at will and is
subject to automatic extension for successive one-year periods. Mr.
Cataldo will be paid $41,666.67 per month during the term of the
Consultant Agreement and will be entitled to participate in the
Company’s bonus plans.
On February 15, 2018, the Company entered into an Executive
Employment Agreement with Mr. Cross, pursuant to which Mr. Cross
will be employed as the Company’s Chief Executive
Officer. The term of the Executive Employment Agreement
is three years and is terminable at will by either the Company or
Mr. Cross and subject to automatic extensions for successive one
year periods. Mr. Cross will be paid an annual salary of
$500,000, paid in equal monthly installment. Mr. Cross is also
entitled to participate in the Company’s bonus plans. Under
the Executive Employment Agreement, the Company has agreed that
it will recommend to the Board that the Company grant Mr.
Cross an option to purchase 2,000,000 shares of the Company’s
common stock at an exercise price equal to the fair market value of
each share as determined by the Board as of the date of the grant.
The stock option grant would vest according to the following
schedule: (i) 34% of the shares on February 15, 2018, (ii) 33% of
the shares on February 15, 2019, and (iii) 33% of the shares on
February 15, 2020.
If any
of our executive officers’ employment with us is terminated
involuntarily, or any executive resigns with good reason as a
result of a change in control, the executive will receive (i) all
compensation and benefits earned through the date of termination of
employment; (ii) a lump-sum payment equal to the greater of (a) the
bonus paid or payable to the executive for the year immediately
prior to the year in which the change in control occurred and (b)
the target bonus under the performance bonus plan in effect
immediately prior to the year in which the change in control
occurs; (iii) a lump-sum payment equivalent to the remaining base
salary (as it was in effect immediately prior to the change in
control) due to the executive from the date of involuntary
termination to the end of the term of the employment agreement or
one half of the executive’s base salary then in effect,
whichever is the greater; and (iv) reimbursement for the cost
of medical, life, disability insurance coverage at a level
equivalent to that provided by us for a period expiring upon the
earlier of (a) one year or (b) the time the executive begins
alternative employment where said insurance coverage is available
and offered to the executive.
5.
Change in Prior Year Financials
The financial statements for the year ended December 31, 2016 have
been restated to correct an error in the reporting of the change in
valuation of the warrant and derivative liabilities. The effect of
the change is to increase the net income for 2016 by
$11,265,000.
On January 22, 2018, the Company entered into a Securities Purchase
Agreement with the investors listed on the Schedule of Buyers
attached thereto (individually, a “Buyer” and
collectively, the “Buyers”) pursuant to which the
Company has agreed to issue to the Buyers senior convertible notes
in an aggregate principal amount of $7,760,510 (the
“Notes”), which Notes shall be convertible into the
Company’s common stock, par value $0.001 per share (the
“Common Stock”), and five-year warrants to purchase the
Company’s Common Stock representing the right to acquire an
aggregate of approximately 1,694,440 shares of Common Stock (the
“Warrants”).
The issuance of the Notes and Warrants is being made in reliance on
the exemption provided by Section 4(a)(2) of the Securities Act of
1933, as amended (the “Securities Act”) for the offer
and sale of securities not involving a public offering, and
Regulation D promulgated under the Securities Act.
Contemporaneously with the execution and delivery of the SPA, the
Company and the Buyers executed and delivered a Registration Rights
Agreement (the “Registration Rights Agreement”)
pursuant to which the Company has agreed to provide certain
registration rights with respect to the Registrable Securities (as
defined thereto) under the 1933 Act and the rules and regulations
promulgated thereunder, and applicable state securities laws. All
descriptions of the SPA, the Registration Rights Agreement, the
Notes and the Warrants contained herein are qualified in their
entirety by reference to the exhibits filed herewith.