UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
| (Mark One) | |
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For
the fiscal year ended:
or
| |
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the transition period from _____________ to _____________
Commission
File Number:
(Exact name of Registrant as specified in its charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
N/A1
(Address of principal executive offices)
(Registrant’s telephone number including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Securities | Trading Symbol(s) | Exchanges on which Registered | ||
Securities registered pursuant to Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days.
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit and post such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| ☒ | Smaller reporting company | ||
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
The
aggregate market value of the registrant’s common stock, $0.001 par value per share, held by non-affiliates on June 30, 2025 was
approximately $
| 1 | Effective as of July 1, 2024, the Company became a fully remote
company. We do not maintain a principal executive office. For purposes of compliance with applicable requirements of the Securities Act
of 1933, as amended, and the Securities Exchange Act of 1934, as amended, any stockholder communication required to be sent to the Company’s
principal executive offices may be directed to |
GT Biopharma, Inc.
FORM 10-K
TABLE OF CONTENTS
| 2 |
SUMMARY RISK FACTORS
Our business involves significant risks. Below is a summary of the material risks that our business faces, which makes an investment in our securities speculative and risky. This summary does not address all these risks. These risks are more fully described below under the heading “Risk Factors” in Part I, Item 1A of this annual report on Form 10-K. Before making investment decisions regarding our securities, you should carefully consider these risks. The occurrence of any of the events or developments described below could have a material adverse effect on our business, results of operations, financial condition, prospects and stock price. In such event, the market price of our securities could decline, and you could lose all or part of your investment. In addition, there are also additional risks not described below that are either not presently known to us or that we currently deem immaterial, and these additional risks could also materially impair our business, operations, or market price of our common stock.
| ● | Our financial condition raises substantial doubt as to our ability to continue as a going concern. | |
| ● | Our business is at an early stage of development and we may not develop therapeutic products that can be commercialized. | |
| ● | We have a history of operating losses and we expect to continue to incur losses for the foreseeable future. We may never generate revenue or achieve profitability. | |
| ● | We will need additional capital to conduct our operations and develop our products, and our ability to obtain the necessary funding is uncertain. | |
| ● | Our research and development costs could exceed our projections requiring us to significantly modify our planned operations. | |
| ● | We have limited clinical testing and regulatory capabilities, and human clinical trials are subject to extensive regulatory requirements, which are very expensive, time-consuming and difficult to design and implement. Our products may fail to achieve necessary safety and efficacy endpoints during clinical trials, which may limit our ability to generate revenues from therapeutic products. | |
| ● | Clinical drug development is costly, time-consuming and uncertain, and we may suffer setbacks in our clinical development program that could harm our business. | |
| ● | If we experience delays or difficulties in the enrollment of patients in clinical trials, those clinical trials could take longer than expected to complete and our receipt of necessary regulatory approvals could be delayed or prevented. | |
| ● | We are subject to extensive regulation, which can be costly and time consuming and can subject us to unanticipated delays. Even if we obtain regulatory approval for some of our products, those products may still face regulatory difficulties. | |
| ● | We will continue to be subject to extensive FDA regulation following any product approvals, and if we fail to comply with these regulations, we may suffer a significant setback in our business. | |
| ● | Our intellectual property may be compromised. | |
| ● | If our efforts to protect the proprietary nature of the intellectual property related to our future product candidates and proprietary technologies are not adequate, we may not be able to compete effectively in our market and our business would be harmed. | |
| ● | Claims that we infringe the intellectual property rights of others may prevent or delay our drug discovery and development efforts. | |
| ● | We may desire, or be forced, to seek additional licenses to use intellectual property owned by third parties, and such licenses may not be available on commercially reasonable terms, or at all. | |
| ● | If we fail to meet our obligations under our license agreements, we may lose our rights to key technologies on which our business depends. | |
| ● | Our reliance on the activities of our non-employee consultants, research institutions and scientific contractors, whose activities are not wholly within our control, may lead to delays in development of our proposed products. |
| 3 |
| ● | Many of our business practices are subject to scrutiny and potential investigation by regulatory and government enforcement authorities, as well as to lawsuits brought by private citizens under federal and state laws. We could become subject to investigations, and our failure to comply with applicable law or an adverse decision in lawsuits may result in adverse consequences to us. If we fail to comply with U.S. healthcare laws, we could face substantial penalties and financial exposure, and our business, operations and financial condition could be adversely affected. | |
| ● | Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any | |
| ● | We may expend our limited resources to pursue a particular product candidate or indication that does not produce any commercially viable products and may fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success. | |
| ● | Our products may be expensive to manufacture, and they may not be profitable if we are unable to control the costs to manufacture them. | |
| ● | We currently lack manufacturing capabilities to produce our therapeutic product candidates at commercial-scale quantities and do not have an alternate manufacturing supply, which would negatively impact our ability to meet any demand for the product. | |
| ● | Our business is based on novel technologies that are inherently expensive and risky and may not be understood by or accepted in the marketplace, which could adversely affect our future value. | |
| ● | We could be subject to product liability lawsuits based on the use of our product candidates in clinical testing or, if obtained, following marketing approval and commercialization. If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to cease clinical testing or limit commercialization of our product candidates. | |
| ● | We rely on third parties to supply candidates for clinical testing and to conduct preclinical and clinical trials of our product candidates. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates. As a result, our business could be substantially harmed. | |
| ● | Our failure to regain or maintain compliance with the Nasdaq Capital Market’s (“Nasdaq”) continued listing requirements could result in the delisting of our common stock. | |
| ● | There has been a limited public market for our common stock, and we do not know whether one will develop to provide adequate liquidity. Furthermore, the trading price for our common stock, should an active trading market develop, may be volatile and could be subject to wide fluctuations in per-share price. | |
| ● | Because our common stock may be deemed a “penny” stock, an investment in our common stock should be considered high-risk and subject to marketability restrictions. |
| 4 |
PART I
CAUTIONARY NOTICE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, including any documents which may be incorporated by reference into this Annual Report, contains “Forward-Looking Statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements other than statements of historical fact are “Forward-Looking Statements” for purposes of these provisions, including our plans of operation, any projections of revenues or other financial items, any statements of the plans and objectives of management for future operations, any statements concerning proposed new products or services, any statements regarding future economic conditions or performance, and any statements of assumptions underlying any of the foregoing. All Forward-Looking Statements included in this document are made as of the date hereof and are based on information available to us as of such date. We assume no obligation to update any Forward-Looking Statement. In some cases, Forward-Looking Statements can be identified by the use of terminology such as “may,” “will,” “expects,” “plans,” “anticipates,” “intends,” “believes,” “estimates,” “potential,” or “continue,” or the negative thereof or other comparable terminology. Although we believe that the expectations reflected in the Forward-Looking Statements contained herein are reasonable, there can be no assurance that such expectations or any of the Forward-Looking Statements will prove to be correct, and actual results could differ materially from those projected or assumed in the Forward-Looking Statements. Future financial condition and results of operations, as well as any Forward-Looking Statements are subject to inherent risks and uncertainties, including any other factors referred to in our press releases and reports filed with the Securities and Exchange Commission. All subsequent Forward-Looking Statements attributable to the company or persons acting on its behalf are expressly qualified in their entirety by these cautionary statements. The factors identified below are believed to be important factors, but not necessarily all of the important factors, that could cause actual results to differ materially from those expressed in any Forward-Looking Statement made by us. Other factors not discussed herein could also have a material adverse effect on us. The following is a list of factors, among others, that could cause actual results to differ materially from those contemplated by the forward-looking statements: our ability to continue as a going concern; our early stage of development and the risk that our product candidates may never be successfully developed; our need for additional capital and uncertainty regarding future financing; the inherent risks, costs and uncertainties of preclinical and clinical development including delays, failures or unexpected safety or efficacy results; our limited regulatory and manufacturing capabilities; our dependence on third parties for research, clinical trials and supply; our reliance on intellectual property and risks related to protecting or licensing those rights; extensive and evolving regulatory requirements; potential product liability claims; risks related to market acceptance and commercialization; volatility in the trading price and limited liquidity of our common stock, and the risk that our failure to comply with Nasdaq listing standards could result in delisting. Additional factors that may have a direct bearing on our operating results are described under “Risk Factors” and elsewhere in this Annual Report on Form 10-K.
Introductory Comment
The corporate predecessor of GT Biopharma, Inc, Diagnostic Data, Inc., was incorporated in the state of California in 1965. Diagnostic Data, Inc. changed its incorporation to the state of Delaware on December 21, 1972 and changed its name to DDI Pharmaceuticals, Inc. on March 11, 1985. On September 7, 1994, DDI Pharmaceuticals, Inc. merged with International BioClinical, Inc. and Bioxytech S.A. and changed its name to OXIS International, Inc. On July 17, 2017, OXIS International, Inc. changed its name to GT Biopharma, Inc.
Throughout this Annual Report on Form 10-K, the terms “GTBP,” “we,” “us,” “our,” “the Company” and “our Company” refer to GT Biopharma, Inc.
The GT Biopharma logo, TriKE®, and other trademarks or service marks of GT Biopharma, Inc. appearing in this quarterly report are the property of the Company. This Annual Report on Form 10-K also contains registered marks, trademarks and trade names of other companies. All other trademarks, registered marks and trade names appearing herein are the property of their respective holders.
The Company is a clinical stage biopharmaceutical company focused on the development and commercialization of novel immune-oncology products based on our proprietary Tri-specific Killer Engager (TriKE®), and Tetra-specific Killer Engager (Dual Targeting TriKE®) platforms. The Company’s TriKE® and Dual Targeting TriKE® platforms generate proprietary therapeutics designed to harness and enhance the cancer killing abilities of a patient’s own natural killer cells (NK cells).
ITEM 1. BUSINESS
We are a clinical stage biopharmaceutical company focused on the development and commercialization of novel immuno-oncology products based on our proprietary Tri-specific Killer Engager (“TriKE®”), and Tetra-specific Killer Engager (“Dual Targeting TriKE®”) fusion protein immune cell engager technology platforms. Our TriKE® and Dual Targeting TriKE® platforms generate proprietary therapeutics designed to harness and enhance the cancer killing abilities of a patient’s own natural killer cells (“NK cells”). Once bound to an NK cell, our moieties are designed to activate the NK cell to direct it to one or more specifically targeted proteins expressed on a specific type of cancer cell or virus infected cell, resulting in the targeted cell’s death. TriKE®s can be designed to target any number of tumor antigens, including B7-H3, HER2, CD33 and PDL1, on hematologic malignancies or solid tumors and do not require patient-specific customization. We believe our TriKE® and Dual Targeting TriKE® platforms that activate endogenous NK cells are potentially safer than T-cell immunotherapy because there is less cytokine release syndrome (“CRS”) and fewer neurological complications. Our preclinical data suggests that this is explained by the TriKE® dependent, CD16 directed, IL-15 proliferation of NK cells, with little effect endogenous T cells.
We are using our TriKE® platform with the intent to bring to market immuno-oncology products that can treat a range of hematologic malignancies, solid tumors, and potentially autoimmune disorders. The platform is scalable, and we are implementing processes to produce investigational new drug (“IND”) ready moieties in a timely manner after a specific TriKE® conceptual design. Specific drug candidates can then be advanced into the clinic on our own or through potential collaborations with partnering companies. We believe our TriKE®s may have the ability, if approved for marketing, to be used as both monotherapy and in combination with other standard-of-care therapies.
Our initial work was conducted in collaboration with the Masonic Cancer Center at the University of Minnesota under a program led by Dr. Jeffrey Miller, Professor of Medicine, and the Interim Director at the Masonic Cancer Center. Dr. Miller, who also serves as our Consulting Senior Medical Director, is a recognized key opinion leader in the field of NK cell and IL-15 biology and their therapeutic potential. We have exclusive rights to the TriKE® platform and are generating additional intellectual property for specific moieties.
| 5 |
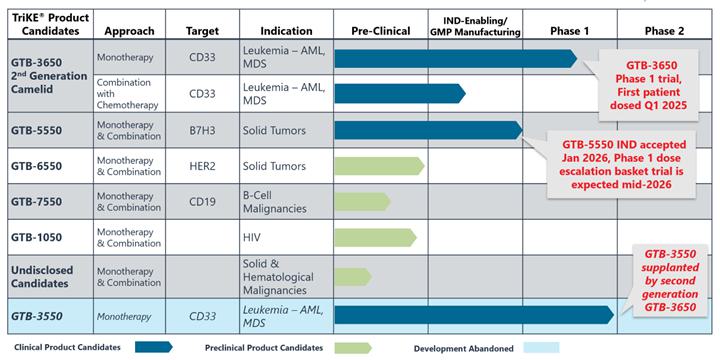
Product Pipeline
Our current TriKE® product candidate pipeline (as of February 23, 2026) is summarized in the table below:
GTB-3550
GTB-3550 was our first TriKE® product candidate and its clinical development was suspended so that we could focus resources on second-generation TriKEs®. GTB-3550 is a tri-specific killer engager, or TriKE, comprised of two single-chain variable fragments (“scFv”) composed of the variable regions of the heavy and light chains of anti-CD16 and anti-CD33 antibodies and a modified form of IL-15. We studied this anti-CD16-IL-15-anti-CD33 TriKE® in CD33 positive leukemias, a marker expressed on tumor cells in acute myelogenous leukemia (“AML”), and myelodysplastic syndrome (“MDS”). The anti-CD33 antibody fragment in GTB-3550 was derived from the M195 humanized anti-CD33 scFv. We believe the approval of the antibody-drug conjugate gemtuzumab validates the targeting of CD33.
We previously announced the interim clinical trial results for GTB-3550, which showed significantly reduced CD 33+ bone marrow blast levels by 33.3%, 61.7%, 63.6%, 50% in Patient 5 (25 µg/kg/day), Patient 7 (50 µg/kg/day), Patient 9 (100 µg/kg/day), and Patient 11 (150 µg/kg/day), respectively. After the end of infusion, GTB-3550 and IL-15 concentrations declined rapidly with overall geometric mean terminal phase elimination half-life (T1/2) of 2.2 and 2.52 hours, respectively. There was minimal CRS resulting from hyperactivation of patient’s T-cell population at doses 5–150 µg/kg/day.
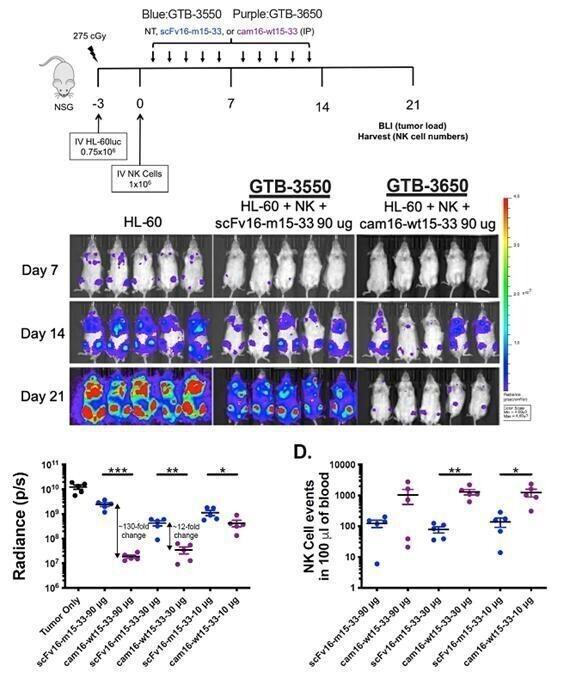
Despite the positive interim clinical trial results, GTB-3550 was replaced by a more potent next-generation camelid nanobody TriKE®, GTB-3650, that similarly targets CD33 on relapsed/refractory AML and high-risk MDS. A key difference between GTB-3550 and GTB-3650 is the incorporation of camelid antibody technology instead of a scFv; our preclinical experience showed markedly enhanced potency of TriKEs® comprised of camelid components. This is illustrated below by better tumor control of AML bearing animals with GTB-3650 (purple dots) compared to GTB-3550 (blue dots). This provided the rationale for pausing further development of GTB-3550 and moving over to solely develop the second-generation, camelid-based TriKE® platform.
| 6 |
Second Generation TriKE®s Utilize Camelid Nanobody Technology
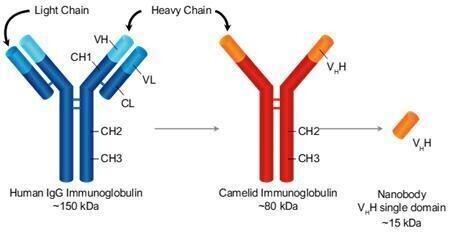
Our goal is to be a leader in immuno-oncology therapies targeting a broad range of indications including hematological malignancies and solid tumors. A key element of our strategy includes introducing a next-generation camelid nanobody platform. Camelid antibodies (often referred to as nanobodies) are smaller than human immunoglobulin, consisting of two heavy chains instead of two heavy and two light chains. These nanobodies have the potential to have greater affinity to target antigens, potentially resulting in greater potency. We are utilizing this camelid antibody structure for all of our new TriKE® product candidates.
To develop second generation TriKE®s, we designed a new humanized CD16 engager derived from a single-domain antibody. While scFvs consist of a heavy and a light variable chain joined by a linker, single-domain antibodies consist of a single variable heavy chain capable of engaging without the need of a light chain counterpart (see figure below).
| 7 |
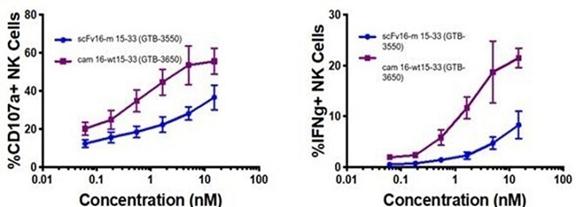
These single-domain antibodies are thought to have certain attractive features for antibody engineering, including physical stability, ability to bind deep grooves, and increased production yields, amongst others. Pre-clinical studies demonstrated increased NK cell activation against CD33+ targets including enhanced NK cell degranulation (% CD107a+) and IFNγ with the single-domain CD16 TriKE® (cam 16-wt15-33; GTB-3650) compared to the original TriKE® (scFv16-m 15-33; GTB-3550) (see figure below). This data was published by Dr. Felices M et al (2020) in Cancer Immunol Res.
CD33+ HL60 Targets in Killing Assays
The purple line represents the GTB-3650 and the blue line represents GTB-3550.
GTB-3650
GTB-3650 is a TriKE® which targets CD33 on the surface of myeloid leukemias and an agonistic camelid engager to the potent activating receptor on NK cells, CD16. Use of this engager enhances the activity of wild type IL-15 included in GTB-3650. The TriKE® approach provides a novel way to specifically target these tumors by leveraging NK cells, which have been shown to mediate relapse protection in this setting, in an anti-CD33-targeted fashion. We are advancing GTB-3650 to clinical studies based on pre-clinical data showing a marked increase in potency compared to GTB-3550, which we anticipate could lead to an enhanced efficacy signal in AML and MDS. We advanced GTB-3650 through requisite preclinical studies and filed an IND application with the U.S. Food and Drug Administration (the “FDA”) in December 2023. In late June 2024, the FDA cleared our IND Application for GTB-3650. We started study enrollment targeting patients with relapsed/refractory AML and high grade MDS on January 21, 2025. This initial study is testing GTB-3650 as monotherapy testing administration 2 weeks on and two weeks off (to prevent NK cell exhaustion) for at least 2 cycles of therapy, as agreed on with the FDA.
| 8 |
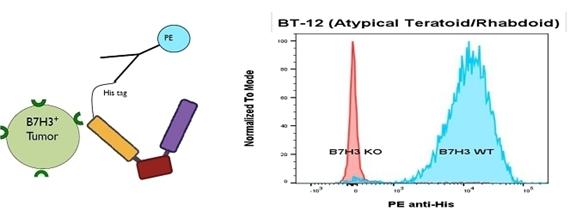
GTB-5550
GTB-5550 is a B7-H3 targeted TriKE® which targets B7-H3 on the surface of advanced solid tumors (figure above). GTB-5550 is our first dual camelid TriKE®. B7-H3 is expressed on a broad spectrum of solid tumor malignancies, allowing our team to target these malignancies through GTB-5550. Pre-clinical work has shown that this molecule has NK-cell targeted activity against a variety of solid tumors, including head and neck cancer squamous cell carcinoma (figure below), prostate cancer, breast cancer, ovarian cancer, glioblastoma, and lung cancer (amongst others).
We advanced GTB-5550 through requisite preclinical studies and filed an IND application with the FDA in October 2023 with a written response from the FDA in December 2023. The main question from the FDA was regarding pre-clinical toxicology and a pivot to subcutaneous dosing. In early January 2026, the FDA cleared our IND Application for GTB-5550. We anticipate starting study enrollment in mid-year 2026. The initial trial is designed as a basket trial for patients with B7-H3+ solid tumors using Monday through Friday dosing (2 weeks on and 2 weeks off to prevent immune exhaustion).
GTB-7550
GTB-7550 TriKE® is a product candidate in development for the treatment of lupus and other autoimmune disorders. GTB-7550 TriKE® is a tri-specific molecule composed of a camelid nanobody that binds the CD16 receptor on NK cells, a scFv engager against CD19 on malignant and normal B cells, and a human IL-15 sequence between them.
Published data shows that GTB-7550 effectively targets CD19+ malignant cell lines and primary chronic lymphocytic leukemia. Preliminary data shows that GTB-7550 can target and eliminate normal B cells, which we are continuing to test in mice. We are currently exploring and assessing potential manufacturers of GTB-7550.
Oncology Markets
Acute Myeloid Leukemia and Myelodysplastic Syndromes
AML is a heterogeneous hematologic stem cell malignancy in adults with an incidence rate of 4.3% per 100,000 populations. The median age at the time of diagnosis is 68 years. AML is an aggressive disease and is fatal without anti-leukemic treatment. AML is the most common form of adult leukemia in the U.S. These patients will require frontline therapy, usually chemotherapy including cytarabine and an anthracycline, a therapy that has not changed in over 40 years. Myelodysplastic syndromes are a heterogeneous group of myeloid neoplasms characterized by dysplastic features of erythroid/myeloid/megakaryocytic lineages, progressive bone marrow failure, a varying percentage of blast cells, and enhanced risk to evolve into acute myeloid leukemia. It is estimated that over 10,000 new cases of MDS are diagnosed each year and there are minimal treatment options; other estimates have put this number higher. In addition, the incidence of MDS is rising for unknown reasons.
| 9 |
B7-H3 Positive Solid Tumors
The B7-H3 protein, which functions as a checkpoint inhibitor, has been identified in many of the most common solid tumor cancers, including but not limited to bladder, breast, cervical, colorectal, endometrial, esophageal, gastric, glioma, kidney, liver, lung, pancreatic, prostate, head and neck cancer, and melanoma. In recent studies, B7-H3 has been identified as a critical promoter of tumor cell proliferation, migration, invasion, epithelial-to-mesenchymal transition, cancer stemness and drug resistance. Because this protein does not seem to be expressed in normal cells, this makes it an attractive target for therapeutic intervention.
Manufacturing
We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of any of our product candidates. We rely on third-party contract manufacturing operations, including Cytovance Biologics, Inc. (“Cytovance”), a related party as of the year ended December 31, 2024, but not as of the year ended December 31, 2025, to produce and/or test our compounds and expect to continue to do so to meet the preclinical and clinical requirements of our potential product candidates as well as for our future commercial needs. We require in our manufacturing and processing agreements that third-party product manufacturers produce intermediates, active pharmaceutical ingredients (“API”), and finished products in accordance with the FDA’s current Good Manufacturing Practices (“cGMP”), and all other applicable laws and regulations. We maintain confidentiality agreements with potential and existing manufacturers to protect our proprietary rights related to our drug candidates.
Competition
The market for therapeutic immuno-oncology products is highly competitive. Our therapeutic immuno-oncology (“IO”) development programs face, and will continue to face, intense competition from pharmaceutical, biopharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies engaged in drug discovery activities or funding, both in the United States and abroad. Some of these competitors are pursuing the development of drugs and other therapies that target the same diseases and conditions that we are targeting with our product candidates.
As a general matter, we also face competition from many companies that are researching and developing cell therapies. Many of these companies have financial and other resources substantially greater than ours. In addition, many of these competitors have significantly greater experience in testing pharmaceutical and other therapeutic products, obtaining FDA and other regulatory approvals, and marketing and selling. If we obtain regulatory approval for any of our product candidates, we also will be competing with respect to manufacturing efficiency and marketing capabilities, areas in which we have limited or no commercial-scale experience. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources’ being concentrated by our competitors. Competition may increase further as a result of advances made in the commercial applicability of our technologies and greater availability of capital for investment in these fields.
TriKE® Patents and Trademarks
On August 24, 2021, two patents were issued by the US Patent Office covering our pipeline of clinical and non-clinical product candidates consisting of tri-specific killer engagers, or TriKE®s, designed to target natural killer, or NK, cells and tumor or virus infected cells forming an immune synapse between the NK cell and the tumor cell thereby inducing NK cell activation at that site. The patents broadly include TriKE®s that target the CD16 receptor, which includes the more potent camelid nanobody sequence, an IL-15 activating domain, and any targeting domain.
University of Minnesota
2023 Sponsored Research Agreement
On May 20, 2024, the Company entered into a sponsored research agreement (the “2023 Sponsored Research Agreement”) with the Regents of the University of Minnesota (the “University of Minnesota”), effective July 1, 2023. Payments totaling approximately $1.7 million were initially due over the life of the agreement. The purpose of the agreement was for the University of Minnesota to continue work with the Company with three major goals in mind: (1) support the Company’s TriKE® product development and commercial GMP manufacturing efforts; (2) TriKE® pharmacokinetics optimization in humans and investigation of effects of altering the route of administration; and (3) research and development of TriKE® platform. The major deliverables proposed were: (1) creation of IND enabling data for TriKE® constructs in support of the Company’s product development and commercial GMP manufacturing efforts outside of the University of Minnesota; (2) TriKE® platform drug delivery changes to allow transition from intravenous (IV) continuous infusion to alternative drug delivery administration (IV bolus, intraperitoneal [IP], subcutaneous [SQ]) and extended PK in humans and gain an increased understanding of changes in the patient’s native NK cell population as a result of alteration of TriKE® administration; and (3) research and development of TriKE® platform combination with other FDA approved (or soon to be approved) therapeutics and alterations to TriKE® platform through formation of immune complexes. Most studies used TriKE® DNA/amino acid sequences created by the Company under existing licensing terms.
On June 18, 2025, the 2023 Sponsored Research Agreement was amended to expire on December 31, 2025. In addition, payments amounting to $216,000 were added bringing the total payments due over the life of the agreement to approximately $1.9 million.
The Company recorded an expense classified as research and development of approximately $863,000 and $1,078,000, pursuant to the 2023 Sponsored Research Agreement, for the years ended December 31, 2025 and 2024, respectively.
As of December 31, 2025, there were no outstanding commitments in relation to unbilled and unaccrued amounts from the University of Minnesota pursuant to the 2023 Sponsored Research Agreement for services that have not yet been rendered as of December 31, 2025.
| 10 |
2016 Exclusive Patent License Agreement
Effective July 18, 2016, the Company entered into an exclusive patent license agreement with the University of Minnesota (as amended, the “2016 Exclusive Patent License Agreement”), to further develop and commercialize cancer therapies using TriKE® technology developed by researchers at the University of Minnesota to target NK cells to cancer. Under the terms of the agreement, the Company receives exclusive rights to conduct research and to develop, make, use, sell, and import TriKE® technology worldwide for the treatment of any disease, state, or condition in humans. The Company is responsible for obtaining all permits, licenses, authorizations, registrations, and regulatory approvals required or granted by any governmental authority anywhere in the world that is responsible for the regulation of products such as the TriKE® technology, including without limitation the FDA and the European Agency for the Evaluation of Medicinal Products in the European Union. The agreement requires an upfront payment of $200,000, and license maintenance fees of $200,000 for years 2017 through 2020, and $100,000 per year beginning in year 2021 and each year thereafter. The agreement also includes 4% royalty fees on the net sales of licensed products, not to exceed 6% under subsequent license agreements or amendments to this agreement, and minimum royalty payments due upon the commencement of commercial sales of licensed product is $250,000 beginning in 2022, $2 million beginning in 2025, and $5 million beginning in 2027 throughout the remainder of the term. The agreement also includes numerous performance milestone payments including clinical development milestone payments totaling $3.1 million, and one-time sales milestone payments of $1 million upon reaching $250 million in cumulative gross sales, and $5 million upon reaching $500 million in cumulative gross sales of licensed products.
Effective May 13, 2024, the Company entered into an amended and restated exclusive patent license agreement with the University of Minnesota (the “A&R 2016 Exclusive Patent License Agreement”). The amendment requires an upfront payment of $145,000 and amends the license maintenance fees to $50,000 in 2025, and $100,000 per year beginning in year 2026 and each year thereafter. The amendment also includes 1% to 5% royalty fees on the net sales of licensed products, not to exceed 6% under subsequent license agreements or amendments, and minimum royalty payments due upon the commencement of commercial sales of licensed product is $250,000 in year one, $2 million in years two through five, and $5 million in year six throughout the remainder of the term. The amendment also includes numerous performance milestone payments including clinical development milestone payments totaling $3.1 million, and one-time sales milestone, and one-time sales milestone payments of $1 million upon reaching $250 million in cumulative gross sales, and $5 million upon reaching $500 million in cumulative gross sales of licensed products.
The Company recorded an expense classified as research and development of $50,000 and $145,000, pursuant to the A&R 2016 Exclusive Patent License Agreement, for the years ended December 31, 2025 and 2024, respectively.
2021 Exclusive License Agreement
Effective March 26, 2021, the Company entered into an exclusive license agreement with the University of Minnesota (the “2021 Exclusive Patent License Agreement”), specific to the B7H3 targeted TriKE®. The agreement requires an upfront payment of $20,000, and license maintenance fees of $5,000 per year beginning in year 2022 and each year thereafter. The agreement also includes 2.5% to 5% royalty fees on the net sales of licensed products, and minimum royalty payments due upon the commencement of commercial sales of licensed product of $250,000 in year one though four, and $2 million beginning in year five and throughout the remainder of the term. The agreement also includes numerous performance milestone payments including clinical development milestone payments totaling $3.1 million, and one-time sales milestone payments of $1 million upon reaching $250 million in cumulative gross sales, and $5 million upon reaching $500 million in cumulative gross sales of licensed products. There is no double payment intended; if one of the milestone payments has been paid under the A&R 2016 Exclusive Patent License Agreement no further payment is due for the corresponding milestone.
The Company recorded an expense classified as research and development of $5,000 and $0, pursuant to the 2021 Exclusive License Agreement, for years ended December 31, 2025 and 2024, respectively.
2024 GTB-3650 Clinical Trial Agreement
On November 18, 2024, the Company entered into an investigator initiated clinical trial agreement (the “2024 Clinical Trial Agreement”) with the University of Minnesota, pursuant to which, the University of Minnesota shall sponsor an IND application for IND 165546 GTB-3650 (the “Research Program”) and shall serve as a sponsor investigator for a phase 1 clinical trial entitled, “GTB-3650 (CD16/IL-15/CD33) Tri-Specific Killer Engager (TriKE) for the Treatment of High Risk Myelodysplastic Syndromes (MDS), Refractory/Relapsed Acute Myeloid Leukemia (AML), and Minimal Residual Disease in AML,” designed by the University of Minnesota (the “Study”). The Research Program is being conducted for clinical research use. The budget for the Study, including without limitations, funding and resources, provides for up to approximately $2 million over the course of three years borne by the Company. The Study data will be owned by the University of Minnesota, however, the Company may use the Study data subject to any applicable signed informed consent documents and authorization forms, applicable law and terms of the 2024 Clinical Trial Agreement. The University of Minnesota and the Company will each have the right to publish the Study results. The 2024 Clinical Trial Agreement may be terminated by the Company or the University of Minnesota at any time upon thirty days’ written notice to the other party, by the University of Minnesota immediately for health, welfare and safety reasons, or by either party if the other party materially breaches the 2024 Clinical Trial Agreement, provided that the breaching party fails to cure such breach within thirty days.
The Company recorded an expense classified as research and development of approximately $750,000 and $190,000, pursuant to the 2024 Clinical Trial Agreement, for the years ended December 31, 2025 and 2024, respectively.
As of December 31, 2025, the Company’s commitments in relation to unbilled and unaccrued amounts from the University of Minnesota pursuant to the 2024 Clinical Trial Agreement for services that have not yet been rendered as of December 31, 2025, amounted to approximately $1.1 million.
| 11 |
Common Stock (February 2024 Reverse Stock-Split)
On February 2, 2024, the Company effectuated a reverse stock-split of its common stock, at a ratio of 1 for 30. The Company’s common stock began trading on a reverse stock-split-adjusted basis on The Nasdaq Capital Market on February 5, 2024, under the existing trading symbol “GTBP.”
As a result of the reverse stock-split, every thirty (30) shares of issued and outstanding common stock were automatically combined into one issued and outstanding share of common stock, without any change in the par value per share. The reverse stock-split reduced the number of shares of common stock outstanding on the effective date of the reverse stock-split from 41,419,000 shares to 1,380,633 shares, subject to minor adjustments due to the treatment of fractional shares.
Proportionate adjustments were made to the per share exercise price and the number of shares of common stock that could be purchased upon exercise of outstanding stock options and warrants for the Company’s common stock, and to the number of shares of common stock reserved for future issuance pursuant to the Company’s 2022 Omnibus Incentive Plan.
All share and per share information within this report have been adjusted to retroactively reflect the reverse stock-split as of the earliest period presented.
The Committed Equity Facility
On May 14, 2025, the Company entered into the common shares purchase agreement (as amended, the “Common Shares Purchase Agreement”) with Bristol Capital, LLC, a Delaware limited liability company (“Bristol”), and Five Narrow Lane, L.P., a Delaware limited partnership (“5NL”), relating to a committed equity facility (the “Committed Equity Facility”), whereby we have the right from time to time at our option to sell to the Facility Investors (as defined below) up to $20 million of our common stock subject to certain conditions and limitations set forth in the Common Shares Purchase Agreement. Bristol subsequently assigned all of its all of its right, title, interest and obligations in the Common Shares Purchase Agreement to its affiliate, Hailstone Peak Funding, LLC, a Delaware limited liability company (“Hailstone,” and together with 5NL, the “Facility Investors”). Sales of the shares of common stock to the Facility Investors under the Common Shares Purchase Agreement, and the timing of any sales, are determined by the Company from time to time in its sole discretion and depend on a variety of factors, including, among other things, market conditions, the trading price of the common stock and determinations by the Company regarding the use of proceeds of such shares of common stock. The net proceeds from any sales under the Common Shares Purchase Agreement will depend on the frequency with, and prices at, which the shares of common stock are sold to the Facility Investors. The purchase price of the shares of common stock that the Company elects to sell to the Facility Investors pursuant to the Common Shares Purchase Agreement are equal to 93% of the volume weighted average price of the shares of Common Stock during the applicable purchase date on which the Company has timely delivered written notice to the Facility Investors directing it to purchase shares of common stock under the Common Shares Purchase Agreement.
Employees and Human Capital Resources
At the date of this Annual Report, we have one full-time employee and numerous consultants to carry on our operations. Many of our activities are outsourced to consultants who provide services to us on a project basis. As business activities require and capital resources permit, we will hire additional employees and consultants to fulfill our Company’s needs.
Available Information
We post our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and any amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, free of charge, on the Investors section of our public website (www.gtbiopharma.com) as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC . In addition, you can read our SEC filings over the Internet at the SEC’s website at www.sec.gov. The contents of these websites are not incorporated into this annual report on Form 10-K. Further, our references to the URLs for these websites are intended to be inactive textual references only.
| 12 |
ITEM 1A. RISK FACTORS
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below in addition to the other information contained in this Annual Report on Form 10-K before deciding whether to invest in shares of our common stock. If any of the following risks actually occur, our business, financial condition or operating results could be harmed. In that case, the trading price of our common stock could decline and you may lose part or all of your investment. In the opinion of management, the risks discussed below represent the material risks known to the company. Additional risks and uncertainties not currently known to us or that we currently deem immaterial may also impair our business, financial condition and operating results and adversely affect the market price of our common stock.
Risks Related to Our Financial Condition and Capital Requirements
Our financial condition raises substantial doubt as to our ability to continue as a going concern.
As of December 31, 2025, we had approximately $6.9 million in cash and cash equivalents and restricted cash, and a working capital of approximately $5.8 million, and we have incurred and expect to continue to incur significant costs in pursuit of our drug candidates. For the year ended December 31, 2025, we recorded a net loss of approximately $28.4 million and used cash in operations of approximately $12.9 million. Our financial statements for the year ended December 31, 2025 have been prepared assuming that we will continue to operate as a going concern, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. To date, we have not generated substantial product revenues from our activities and have incurred substantial operating losses. We expect that we will continue to generate substantial operating losses for the foreseeable future until we complete development and approval of our product candidates. We expect to continue to fund our operations primarily through utilization of our current financial resources and additional raises of capital.
These conditions raise substantial doubt about our ability to continue as a going concern. In addition, the Company’s independent registered public accounting firm, in its report on the Company’s December 31, 2025 financial statements, raised substantial doubt about the Company’s ability to continue as a going concern. The Company has evaluated the significance of the uncertainty regarding the Company’s financial condition in relation to its ability to meet its obligations, which has raised substantial doubt about the Company’s ability to continue as a going concern. While it is very difficult to estimate the Company’s future liquidity requirements, the Company believes if it is unable to obtain additional financing, existing cash resources will not be sufficient to enable it to fund the anticipated level of operations through one year from the date the accompanying financial statements are issued. There can be no assurances that the Company will be able to secure additional financing on acceptable terms or that it will provide us with sufficient funds to meet our objectives. In the event the Company does not secure additional financing, the Company will be forced to delay, reduce, or eliminate some or all of its discretionary spending, which could adversely affect the Company’s business prospects, ability to meet long-term liquidity needs and the ability to continue operations.
Our business is at an early stage of development and we may not develop therapeutic products that can be commercialized.
Our business is at an early stage of development. We do not have immune-oncology products in late-stage clinical trials. We are still in the early stages of identifying and conducting research on potential therapeutic products. Our potential therapeutic products will require significant research and development and pre-clinical and clinical testing prior to regulatory approval in the United States and other countries. We may not be able to obtain regulatory approvals, enter clinical trials for any of our product candidates, or commercialize any products for years, if at all. Our product candidates may prove to have undesirable and unintended side effects or other characteristics adversely affecting their safety, efficacy or cost effectiveness that could prevent or limit their use. Any product using any of our technology may fail to provide the intended therapeutic benefits or achieve therapeutic benefits equal to or better than the standard of treatment at the time of testing or production.
We have a history of operating losses and we expect to continue to incur losses for the foreseeable future and we may never generate revenue or achieve profitability.
During the year ended December 31, 2025, the Company reported a net loss of approximately $28.4 million and as of December 31, 2025, and had an accumulated deficit of approximately $724 million. We have not generated any revenue to date and are not profitable, and have incurred losses in each year since our inception. We do not expect to generate any product sales or royalty revenues for the foreseeable future. We expect to continue to incur significant additional operating losses for the foreseeable future as we expand research and development and clinical trial efforts.
Our ability to achieve long-term profitability is dependent upon obtaining regulatory approvals for our products and successfully commercializing our products alone or with third parties. However, our operations may not be profitable even if any of our products under development are successfully developed and produced and thereafter commercialized. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
Even if we succeed in commercializing one or more of our product candidates, we expect to continue to incur substantial research and development and other expenditures to develop and market additional product candidates. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenue. Our prior losses and expected future losses have had and will continue to have an adverse effect on our stockholders’ equity and working capital.
| 13 |
We will need additional capital to conduct our operations and develop our products, and our ability to obtain the necessary funding is uncertain.
We have used a significant amount of cash since inception to finance the continued development and testing of our product candidates, and we expect to need substantial additional capital resources to develop our product candidates going forward and launch and commercialize any product candidates for which we receive regulatory approval.
We may not be successful in generating and/or maintaining operating cash flow, and the timing of our capital expenditures and other expenditures may not result in cash sufficient to sustain our operations through the commercialization of our product candidates. If financing is not sufficient and additional financing is not available or available only on terms that are detrimental to our long-term survival, it could have a material adverse effect on our ability to continue to operate. The timing and degree of any future capital requirements will depend on many factors, including:
| ● | the accuracy of the assumptions underlying our estimates for capital needs in 2026 and beyond; |
| ● | scientific and clinical progress in our research and development programs; |
| ● | the magnitude and scope of our research and development programs and our ability to establish, enforce and maintain strategic arrangements for research, development, clinical testing, manufacturing and marketing; |
| ● | our progress with pre-clinical development and clinical trials; |
| ● | the time and costs involved in obtaining regulatory approvals; |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims; and |
| ● | the number and type of product candidates that we pursue. |
Additional financing through strategic collaborations, public or private equity or debt financings or other financing sources may not be available on acceptable terms, or at all. Additional equity financing could result in significant dilution to our stockholders, and any debt financings will likely involve covenants restricting our business activities. Further, if we obtain additional funds through arrangements with collaborative partners, these arrangements may require us to relinquish rights to some of our technologies, product candidates or products that we would otherwise seek to develop and commercialize on our own.
If sufficient capital is not available, we may be required to delay, reduce the scope of or eliminate one or more of our research or product development initiatives, any of which could have a material adverse effect on our financial condition or business prospects. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. Attempting to secure additional financing our product candidates.
Our research and development costs could exceed our projections requiring us to significantly modify our planned operations.
The actual cost of our research and development programs could differ significantly from our current projections if we change our planned development process. In the event that actual costs of our clinical program, or any of our other ongoing research activities, are significantly higher than our current estimates, we may be required to significantly modify our planned level of operations.
The successful development of any product candidate is highly uncertain. It is difficult to reasonably estimate or know the nature, timing and costs of the efforts necessary to complete the development of, or the period in which material net cash inflows are expected to commence from any product candidate, due to the numerous risks and uncertainties associated with developing drugs. Any failure to complete any stage of the development of products in a timely manner could have a material adverse effect on our operations, financial position and liquidity.
| 14 |
Risks Related to Clinical Development and Potential Regulatory Approval
We have limited clinical testing and regulatory capabilities, and human clinical trials are subject to extensive regulatory requirements, which are very expensive, time-consuming and difficult to design and implement. Our products may fail to achieve necessary safety and efficacy endpoints during clinical trials, which may limit our ability to generate revenues from therapeutic products.
We cannot assure that we will be able to invest or develop resources for clinical trials successfully or as expediently as necessary. In particular, human clinical trials can be very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is time consuming. We estimate that clinical trials of our product candidates will take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be affected by several factors, including:
| ● | unforeseen safety issues; | |
| ● | determination of dosing issues; | |
| ● | inability to demonstrate effectiveness during clinical trials; | |
| ● | slower than expected rates of patient recruitment; | |
| ● | inability to monitor patients adequately during or after treatment; and | |
| ● | inability or unwillingness of medical investigators to follow our clinical protocols. |
In addition, we or the FDA, may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our investigational new drug application, or IND, submissions or the conduct of these trials.
Clinical drug development is costly, time-consuming and uncertain, and we may suffer setbacks in our clinical development program that could harm our business.
Clinical drug development for our product candidates is costly, time-consuming and uncertain. Our product candidates are in various stages of development and while we expect that clinical trials for these product candidates will continue for several years, such trials may take significantly longer than expected to complete. In addition, we, the FDA, an Institutional Review Board (“IRB”), or other regulatory authorities, including state and local agencies and counterpart agencies in foreign countries, may suspend, delay, require modifications to or terminate our clinical trials at any time, for various reasons, including:
| ● | discovery of safety or tolerability concerns, such as serious or unexpected toxicities or side effects or exposure to otherwise unacceptable health risks, with respect to study participants; | |
| ● | lack of effectiveness of any product candidate during clinical trials or the failure of our product candidates to meet specified endpoints; | |
| ● | delays in subject recruitment and enrollment in clinical trials or inability to enroll a sufficient number of patients in clinical trials to ensure adequate statistical ability to detect statistically significant treatment effects; | |
| ● | difficulty in retaining subjects and volunteers in clinical trials; | |
| ● | difficulty in obtaining the IRB’s approval for studies to be conducted at each clinical trial site; | |
| ● | inadequacy of or changes in our manufacturing process or the product formulation or method of delivery; | |
| ● | delays or failure in reaching agreement on acceptable terms in clinical trial contracts or protocols with prospective Contract Research Organizations, (“CROs”), clinical trial sites and other third-party contractors; | |
| ● | inability to add a sufficient number of clinical trial sites; | |
| ● | uncertainty regarding proper formulation and dosing; | |
| ● | failure by us, our employees, our CROs or their employees or other third-party contractors to comply with contractual and applicable regulatory requirements or to perform their services in a timely or acceptable manner; | |
| ● | scheduling conflicts with participating clinicians and clinical institutions; | |
| ● | failure to design appropriate clinical trial protocols; | |
| ● | inability or unwillingness of medical investigators to follow our clinical protocols; | |
| ● | difficulty in maintaining contact with subjects during or after treatment, which may result in incomplete data; or | |
| ● | changes in applicable laws, regulations and regulatory policies. |
It may take longer to complete our clinical trials than we project, or we may not be able to complete them at all.
For budgeting and planning purposes, we have projected the date for the commencement, continuation and completion of our various clinical trials. However, a number of factors, including scheduling conflicts with participating clinicians and clinical institutions, and difficulties in identifying and enrolling patients who meet trial eligibility criteria, may cause significant delays. We may not commence or complete clinical trials involving any of our products as projected or may not conduct them successfully.
We expect to rely on medical institutions, academic institutions or clinical research organizations to conduct, supervise or monitor some or all aspects of clinical trials involving our products. We will have less control over the timing and other aspects of these clinical trials than if we conducted them entirely on our own. If we fail to commence or complete, or experience delays in, any of our planned clinical trials, our stock price and our ability to conduct our business as currently planned could be harmed.
| 15 |
If we experience delays or difficulties in the enrollment of patients in clinical trials, those clinical trials could take longer than expected to complete and our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials for our product candidates if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA, or similar regulatory authorities outside the United States. In particular, because we are focused on patients with molecularly defined cancers, our pool of suitable patients may be smaller and more selective and our ability to enroll a sufficient number of suitable patients may be limited or take longer than anticipated. In addition, some of our competitors have ongoing clinical trials for product candidates that treat the same indications as our product candidates, and patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’ product candidates.
Patient enrollment for any of our clinical trials may also be affected by other factors, including without limitation:
| ● | the severity of the disease under investigation; |
| ● | the frequency of the molecular alteration we are seeking to target in the applicable trial; |
| ● | the eligibility criteria for the study in question; |
| ● | the perceived risks and benefits of the product candidate under study; |
| ● | the extent of the efforts to facilitate timely enrollment in clinical trials; |
| ● | the patient referral practices of physicians; |
| ● | the ability to monitor patients adequately during and after treatment; and |
| ● | the proximity and availability of clinical trial sites for prospective patients. |
Our inability to enroll a sufficient number of patients for our clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for our product candidates, and we may not have or be able to obtain sufficient cash to fund such increased costs when needed, which could result in the further delay or termination of the trial.
Consistent with our general product development strategy, we intend to design future trials for our product candidates to include some patients with the applicable clinical characteristics, stage of therapy, molecular alterations, biomarkers, and/or cell surface antigens that determine therapeutic options, or are indicators of the disease, with a view to assessing possible early evidence of potential therapeutic effect. If we are unable to locate and include such patients in those trials, then our ability to make those early assessments and to seek participation in FDA expedited review and approval programs, including breakthrough therapy and fast track designation, or otherwise to seek to accelerate clinical development and regulatory timelines, could be compromised.
We are subject to extensive regulation, which can be costly and time consuming and can subject us to unanticipated delays. Even if we obtain regulatory approval for some of our products, those products may still face regulatory difficulties.
All of our potential products, processing and manufacturing activities, are subject to comprehensive regulation by the FDA in the United States and by comparable authorities in other countries. The process of obtaining FDA and other required regulatory approvals, including foreign approvals, is expensive and often takes many years and can vary substantially based upon the type, complexity and novelty of the products involved. In addition, regulatory agencies may lack experience with our technologies and products, which may lengthen the regulatory review process, increase our development costs and delay or prevent their commercialization.
If we violate regulatory requirements at any stage, whether before or after we obtain marketing approval, the FDA may take enforcement action(s) against us, which could include issuing a warning or untitled letter, placing a clinical hold on an ongoing clinical trial, product seizure, enjoining our operations, refusal to consider our applications for pre-market approval, refusal of an investigational new drug application, fines, or even civil or criminal liability, any of which could materially harm our reputation and financial results. Additionally, we may not be able to obtain the labeling claims necessary or desirable for the promotion of our products. We may also be required to undertake post marketing trials to provide additional evidence of safety and effectiveness. In addition, if we or others identify side effects after any of our adoptive therapies are on the market, or if manufacturing problems occur, regulators may withdraw their approval and reformulations, additional clinical trials, changes in labeling of our products, and additional marketing applications may be required.
| 16 |
Any of the following factors, among others, could cause regulatory approval for our product candidates to be delayed, limited or denied:
| ● | the product candidates require significant clinical testing to demonstrate safety and effectiveness before applications for marketing approval can be filed with the FDA and other regulatory authorities; |
| ● | data obtained from pre-clinical and nonclinical animal testing and clinical trials can be interpreted in different ways, and regulatory authorities may not agree with our respective interpretations or may require us to conduct additional testing; |
| ● | negative or inconclusive results or the occurrence of serious or unexpected adverse events during a clinical trial could cause us to delay or terminate development efforts for a product candidate; and/or |
| ● | FDA and other regulatory authorities may require expansion of the size and scope of the clinical trials. |
Any difficulties or failures that we encounter in securing regulatory approval for our product candidates would likely have a substantial adverse impact on our ability to generate product sales, and could make a search for a collaborative partner more difficult.
Obtaining regulatory approval even after clinical trials that are believed to be successful is an uncertain process.
Even if we complete our planned clinical trials and believe the results were successful, obtaining regulatory approval is a lengthy, expensive and uncertain process, and the FDA or other regulatory agencies may delay, limit or deny approval of any of our applications for pre-market approval for many reasons, including:
| ● | we may not be able to demonstrate to the FDA’s satisfaction that our product candidates are safe and effective for any indication; |
| ● | the results of clinical trials may not meet the level of statistical significance or clinical significance required by the FDA for approval; |
| ● | the FDA may disagree with the number, design, size, conduct or implementation of our clinical trials; |
| ● | the FDA may not find the data from pre-clinical studies and clinical trials sufficient to demonstrate that the clinical and other benefits of our product candidates outweigh their safety risks; |
| ● | the FDA may disagree with our interpretation of data from pre-clinical studies or clinical trials, or may not accept data generated at our clinical trial sites; |
| ● | the data collected from pre-clinical studies and clinical trials of our product candidates may not be sufficient to support the submission of applications for regulatory approval; |
| ● | the FDA may have difficulties scheduling an advisory committee meeting in a timely manner, or the advisory committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional pre-clinical studies or clinical trials, limitations on approved labeling, or distribution and use restrictions; |
| ● | the FDA may require development of a risk evaluation and mitigation strategy as a condition of approval; |
| ● | the FDA may identify deficiencies in the manufacturing processes or facilities of third-party manufacturers with which we enter into agreements for clinical and commercial supplies; |
| ● | the FDA may change their approval policies or adopt new regulations that adversely affect our applications for pre-market approval; and |
| ● | the FDA may require simultaneous approval for both adults and for children and adolescents delaying needed approvals, or we may have successful clinical trial results for adults but not children and adolescents, or vice versa. |
Before we can submit an application for regulatory approval in the United States, we must conduct a pivotal, registrational trial. We will also need to agree on a protocol with the FDA for a clinical trial before commencing the trial. Registrational clinical trials frequently produce unsatisfactory results even though prior clinical trials were successful. Therefore, even if the results of our early phase trials are successful, the results of the additional trials that we conduct may or may not be successful. Further, our product candidates may not be approved even if they achieve their primary endpoints in registrational clinical trials. The FDA or other foreign regulatory authorities may disagree with our trial design and our interpretation of data from preclinical studies and clinical trials. Any of these regulatory authorities may change requirements for the approval of a product candidate even after reviewing and providing comments or advice on a protocol for a clinical trial. The FDA or other regulatory agencies may require that we conduct additional clinical, nonclinical, manufacturing validation or drug product quality studies and submit those data before considering or reconsidering the application. Depending on the extent of these or any other studies, approval of any applications that we submit may be delayed by several years, or may require us to expend more resources than we have available. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the FDA or other regulatory agencies.
| 17 |
In addition, the FDA or other regulatory agencies may also approve a product candidate for fewer or more limited indications than we request, may impose significant limitations related to use restrictions for certain age groups, warnings, precautions or contraindications or may grant approval contingent on the performance of costly post-marketing clinical trials or risk mitigation requirements.
We will continue to be subject to extensive FDA regulation following any product approvals, and if we fail to comply with these regulations, we may suffer a significant setback in our business.
Even if we are successful in obtaining regulatory approval of our product candidates, we will continue to be subject to the requirements of and review by, the FDA and comparable regulatory authorities in the areas of manufacturing processes, post-approval clinical data, adverse event reporting, labeling, advertising and promotional activities, among other things. In addition, any marketing approval we receive may be limited in terms of the approved product indication or require costly post-marketing testing and surveillance. Discovery after approval of previously unknown problems with a product, manufacturer or manufacturing process, or a failure to comply with regulatory requirements, may result in enforcement actions such as:
| ● | warning letters or other actions requiring changes in product manufacturing processes or restrictions on product marketing or distribution; |
| ● | product recalls or seizures or the temporary or permanent withdrawal of a product from the market; |
| ● | suspending any ongoing clinical trials; |
| ● | temporary or permanent injunctions against our production operations; |
| ● | refusal of our applications for pre-market approval or an investigational new drug application; and |
| ● | fines, restitution or disgorgement of profits or revenue, the imposition of civil penalties or criminal prosecution. |
The occurrence of any of these actions would likely cause a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Intellectual Property
Our intellectual property may be compromised.
Part of our value going forward depends on the intellectual property rights that we have been and are acquiring. There may have been many persons involved in the development of our intellectual property, and we seek to protect our intellectual property, in part, by entering into confidentiality agreements with parties who have access. Despite these efforts, we may not be successful in obtaining the necessary rights from all of them or these parties may breach the agreements and disclose our proprietary information. In addition, it is possible that in the future, third parties may challenge our intellectual property rights. We may not be successful in protecting our intellectual property rights. In either event, we may lose the value of our intellectual property, and if so, our business prospects may suffer.
If our efforts to protect the proprietary nature of the intellectual property related to our future product candidates and proprietary technologies are not adequate, we may not be able to compete effectively in our market and our business would be harmed.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our future product candidates and proprietary technologies. Any disclosure to or misappropriation by third parties of our trade secret or other confidential information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding any competitive advantage we may derive from this information.
The strength of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and can be uncertain. The patent applications we own or license may fail to result in issued patents in the United States or in foreign countries. Third parties may challenge the validity, enforceability or scope of any issued patents we own or license or any applications that may be issued as patents in the future, which may result in those patents being narrowed, invalidated or held unenforceable. Even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from developing similar products that do not fall within the scope of our patents. If the breadth or strength of protection provided by the patents we hold or pursue is threatened, our ability to commercialize any product candidates with technology protected by those patents could be threatened. Further, if we encounter delays in our clinical trials, the time during which we would have patent protection for any covered product candidates that obtain regulatory approval would be reduced. Since patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain at the time of filing that we are the first to file any patent application related to our product candidates.
| 18 |
In addition to the protection afforded by patents, we seek to rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our discovery platform and drug development processes that involve proprietary know-how, information or technology that is not covered by patents or not amenable to patent protection. Although we require all of our employees and certain consultants and advisors to assign inventions to us, and all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, our trade secrets and other proprietary information may be disclosed or competitors may otherwise gain access to such information or independently develop substantially equivalent information. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant difficulty in protecting and defending our intellectual property both in the United States and abroad. If we are unable to prevent material disclosure of the trade secret intellectual property related to our technologies to third parties, we may not be able to establish or maintain the competitive advantage that we believe is provided by such intellectual property, which could materially adversely affect our market position and business and operational results.
Claims that we infringe the intellectual property rights of others may prevent or delay our drug discovery and development efforts.
Our research, development and commercialization activities, as well as any product candidates or products resulting from those activities, may infringe or be accused of infringing a patent or other form of intellectual property under which we do not hold a license or other rights. Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents of which we are currently unaware, with claims that cover the use or manufacture of our product candidates or the practice of our related methods. Because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes one or more claims of these patents. If our activities or product candidates infringe the patents or other intellectual property rights of third parties, the holders of such intellectual property rights may be able to block our ability to commercialize such product candidates or practice our methods unless we obtain a license under the intellectual property rights or until any applicable patents expire or are determined to be invalid or unenforceable.
Defense of any intellectual property infringement claims against us, regardless of their merit, would involve substantial litigation expense and would be a significant diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, obtain one or more licenses from third parties, limit our business to avoid the infringing activities, pay royalties and/or redesign our infringing product candidates or methods, any or all of which may be impossible or require substantial time and monetary expenditure. Further, if we were to seek a license from the third-party holder of any applicable intellectual property rights, we may not be able to obtain the applicable license rights when needed or on commercially reasonable terms, or at all. The occurrence of any of the above events could prevent us from continuing to develop and commercialize one or more of our product candidates and our business could materially suffer.
We may desire, or be forced, to seek additional licenses to use intellectual property owned by third parties, and such licenses may not be available on commercially reasonable terms or at all.
A third party may hold intellectual property, including patent rights, that are important or necessary to the development of our product candidates, in which case we would need to obtain a license from that third party or develop a different formulation of the product that does not infringe upon the applicable intellectual property, which may not be possible. Additionally, we may identify product candidates that we believe are promising and whose development and other intellectual property rights are held by third parties. In such a case, we may desire to seek a license to pursue the development of those product candidates. Any license that we may desire to obtain or that we may be forced to pursue may not be available when needed on commercially reasonable terms or at all. Any inability to secure a license that we need or desire could have a material adverse effect on our business, financial condition and prospects.
The patent protection covering some of our product candidates may be dependent on third parties, who may not effectively maintain that protection.
While we expect that we will generally seek to gain the right to fully prosecute any patents covering product candidates we may in-license from third-party owners, there may be instances when platform technology patents that cover our product candidates remain controlled by our licensors. If any of our current or future licensing partners that retain the right to prosecute patents covering the product candidates we license from them fail to appropriately maintain that patent protection, we may not be able to prevent competitors from developing and selling competing products or practicing competing methods and our ability to generate revenue from any commercialization of the affected product candidates may suffer.
| 19 |
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe our patents or the patents of our current or potential licensors. To attempt to stop infringement or unauthorized use, we may need to enforce one or more of our patents, which can be expensive and time-consuming and distract management. If we pursue any litigation, a court may decide that a patent of ours or our licensor’s is not valid or is unenforceable, or may refuse to stop the other party from using the relevant technology on the grounds that our patents do not cover the technology in question. Further, the legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, which could reduce the likelihood of success of any infringement proceeding we pursue in any such jurisdiction. An adverse result in any infringement litigation or defense proceedings could put one or more of our patents at risk of being invalidated, held unenforceable, or interpreted narrowly and could put our patent applications at risk of not issuing, which could limit our ability to exclude competitors from directly competing with us in the applicable jurisdictions.
Interference proceedings provoked by third parties or brought by the U.S. Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to use it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms, or at all. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees.
If we are unsuccessful in obtaining or maintaining patent protection for intellectual property in development, our business and competitive position would be harmed.
We are seeking patent protection for some of our technology and product candidates. Patent prosecution is a challenging process and is not assured of success. If we are unable to secure patent protection for our technology and product candidates, our business may be adversely impacted.
In addition, issued patents and pending international applications require regular maintenance. Failure to maintain our portfolio may result in loss of rights that may adversely impact our intellectual property rights, for example by rendering issued patents unenforceable or by prematurely terminating pending international applications.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and product candidates, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We currently, and expect in the future to continue to, seek to protect these trade secrets, in part, by entering into confidentiality agreements with parties who have access to them, such as our employees, collaborators, contract manufacturers, consultants, advisors and other third parties. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for any such disclosure. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they disclose the trade secrets, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
If we fail to meet our obligations under our license agreements, we may lose our rights to key technologies on which our business depends.
Our business depends in part on licenses from third parties. These third-party license agreements impose obligations on us, such as payment obligations and obligations to diligently pursue development of commercial products under the licensed patents. If a licensor believes that we have failed to meet our obligations under a license agreement, the licensor could seek to limit or terminate our license rights, which could lead to costly and time-consuming litigation and, potentially, a loss of the licensed rights. During the period of any such litigation, our ability to carry out the development and commercialization of potential products could be significantly and negatively affected. If our license rights were restricted or ultimately lost, our ability to continue our business based on the affected technology platform could be severely adversely affected.
| 20 |
Risks Related to Our Business Operations and Industry
If we lose key personnel, or if we cannot recruit qualified additional employees to carry on our business operations, we may not be able to implement our business strategy.
We currently have one full-time employee and numerous consultants to carry on our operations. The loss of the services of any of our employees or consultants including, in particular our Chief Executive Officer and Executive Chairman of the Board, could delay our product development programs and our research and development efforts. In order to develop our business in accordance with our business strategy, we will have to hire additional qualified personnel, including in the areas of manufacturing, clinical trials management, regulatory affairs, finance, discovery biology, and business development. We will need to raise sufficient funds to hire and retain the necessary employees and consultants.
Moreover, because of the specialized nature of our business, we are highly dependent on our ability to identify, hire, train and retain highly qualified scientific and technical personnel for the research and development activities we conduct or sponsor. The loss of one or more key executive officers, scientific or operational team members would be significantly detrimental to us. In addition, recruiting and retaining qualified scientific personnel to perform research and development work is critical to our success. Our anticipated growth and expansion into areas and activities requiring additional expertise, such as discovery biology, clinical testing, regulatory compliance, manufacturing and compliance, will require the addition of new management personnel and the development of additional expertise by existing management personnel.
There is intense competition for a limited number of qualified personnel among biopharmaceutical, biotechnology, pharmaceutical and other businesses. Many of the other pharmaceutical companies against which we compete for qualified personnel have greater financial and other resources, different risk profiles, longer histories in the industry and greater ability to provide valuable cash or stock incentives to potential recruits than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high quality candidates than what we are able to offer as an early-stage company. If we are unable to continue to attract and retain high quality personnel, the rate and success at which we can develop and commercialize product candidates will be limited.
We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
In the past, many of our employees were employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, with contractual provisions and other procedures, we may be subject to claims that these employees or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employers. Litigation may be necessary to defend against any such claims.
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact contributes to the development of intellectual property that we regard as our own. Further, the terms of such assignment agreements may be breached and we may not be able to successfully enforce their terms, which may force us to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of intellectual property rights we may regard and treat as our own.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause our business to suffer.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with regulations of governmental authorities, such as the U.S. Food and Drug Administration (“FDA”) or the European Medicines Agency (“EMA”), to provide accurate information to the FDA or EMA, to comply with manufacturing standards we have established, to comply with federal, state and international healthcare fraud and abuse laws and regulations as they may become applicable to our operations, to report financial information or data accurately or to disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained during clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we currently take and the procedures we may establish in the future as our operations and employee base expand to detect and prevent this type of activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure by our employees to comply with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
Our reliance on the activities of our non-employee consultants, research institutions and scientific contractors, whose activities are not wholly within our control, may lead to delays in development of our proposed products.
We rely extensively upon and have relationships with scientific consultants at academic and other institutions, some of whom conduct research at our request, and other consultants with expertise in clinical development strategy or other matters. These consultants are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. We have limited control over the activities of these consultants and, except as otherwise required by our collaboration and consulting agreements to the extent they exist, can expect only limited amounts of their time to be dedicated to our activities. These research facilities may have commitments to other commercial and non-commercial entities. We have limited control over the operations of these laboratories and can expect only limited amounts of time to be dedicated to our research goals.
| 21 |
Many of our business practices are subject to scrutiny and potential investigation by regulatory and government enforcement authorities, as well as to lawsuits brought by private citizens under federal and state laws. We could become subject to investigations, and our failure to comply with applicable law or an adverse decision in lawsuits may result in adverse consequences to us. If we fail to comply with U.S. healthcare laws, we could face substantial penalties and financial exposure, and our business, operations and financial condition could be adversely affected.
While payment is not yet available from third-party payors (government or commercial) for our product, our goal is to obtain such coverage as soon as possible after product approval and commercial launch in the U.S. If this occurs, the availability of such payment would mean that many healthcare laws would place limitations and requirements on the manner in which we conduct our business (including our sales and promotional activities and interactions with healthcare professionals and facilities) and could result in liability and exposure to us. In some instances, our interactions with healthcare professionals and facilities that occurred prior to commercialization could have implications at a later date. The laws that may affect our ability to operate include, among others: (i) the federal healthcare programs Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as Medicare or Medicaid; (ii) federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent, and which may apply to entities like us under theories of “implied certification” where the government and qui tam relators may allege that device companies are liable where a product that was paid for by the government in whole or in part was promoted “off-label,” lacked necessary approval, or failed to comply with good manufacturing practices or other laws; (iii) transparency laws and related reporting and/or disclosures such as the Sunshine Act; and/or (iv) state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, many of which differ from their federal counterparts in significant ways, thus complicating compliance efforts.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, exclusion from participation in government healthcare programs, damages, fines and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. The risk of our being found in violation of these laws is increased by the fact that their provisions are open to a variety of evolving interpretations and enforcement discretion. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
Both federal and state government agencies have heightened civil and criminal enforcement efforts. There are numerous ongoing investigations of healthcare pharmaceutical companies and others in the healthcare space, as well as their executives and managers. In addition, amendments to the Federal False Claims Act, have made it easier for private parties to bring qui tam (whistleblower) lawsuits against companies under which the whistleblower may be entitled to receive a percentage of any money paid to the government. In addition, the Affordable Care Act amended the federal civil False Claims Act to provide that a claim that includes items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Penalties include substantial fines for each false claim, plus three times the amount of damages that the federal government sustained because of the act of that person or entity and/or exclusion from the Medicare program. In addition, a majority of states have adopted similar state whistleblower and false-claims provision. There can be no assurance that our activities will not come under the scrutiny of regulators and other government authorities or that our practices will not be found to violate applicable laws, rules and regulations or prompt lawsuits by private citizen “relators” under federal or state false claims laws. Any future investigations of our business or executives, or enforcement action or prosecution, could cause us to incur substantial costs, and result in significant liabilities or penalties, as well as damage to our reputation.
Laws impacting the U.S. healthcare system are subject to a great deal of uncertainty, which may result in adverse consequences to our business.
There have been a number of legislative and regulatory proposals to change the healthcare system, reduce the costs of healthcare and change medical reimbursement policies. Doctors, clinics, hospitals and other users of our products may decline to purchase our products to the extent there is uncertainty regarding coverage from government or commercial payors. Further proposed legislation, regulation and policy changes affecting third-party reimbursement are likely. Among other things, Congress has in the past proposed changes to and the repeal of the Patient Protection and Affordable Care and Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the “Affordable Care Act”), and lawsuits have been brought challenging aspects of the law at various points. There have been repeated recent attempts by Congress to repeal or replace the Affordable Care Act. At this time, it remains unclear whether there will be any changes made to or any repeal or replacement of the Affordable Care Act, with respect to certain of its provisions or in its entirety. We are unable to predict what legislation or regulation, if any, relating to the health care industry or third-party coverage and reimbursement may be enacted in the future at the state or federal level, or what effect such legislation or regulation may have on us. Denial of coverage and reimbursement of our products, or the revocation or changes to coverage and reimbursement policies, could have a material adverse effect on our business, results of operations and financial condition.
| 22 |
We may not be successful in our efforts to build a pipeline of product candidates.
A key element of our strategy is to use and expand our product platform to build a pipeline of product candidates and progress those product candidates through clinical development for the treatment of a variety of different types of cancer. Even if we are successful in building a product pipeline, the potential product candidates that we identify may not be suitable for clinical development for a number of reasons, including causing harmful side effects or demonstrating other characteristics that indicate a low likelihood of receiving marketing approval or achieving market acceptance. If our methods of identifying potential product candidates fail to produce a pipeline of potentially viable product candidates, then our success as a business will be dependent on the success of fewer potential product candidates, which introduces risks to our business model and potential limitations to any success we may achieve.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Additionally, if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may withdraw approvals of such product; |
| ● | regulatory authorities may require additional warnings on the product’s label; |
| ● | we may be required to create a medication guide for distribution to patients that outlines the risks of such side effects; |
| ● | we could be sued and held liable for harm caused to patients; and |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
We may expend our limited resources to pursue a particular product candidate or indication that does not produce any commercially viable products and may fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we must focus our efforts on particular research programs and product candidates for specific indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Further, our resource allocation decisions may result in our use of funds for research and development programs and product candidates for specific indications that may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. Any such failure to improperly assess potential product candidates could result in missed opportunities and/or our focus on product candidates with low market potential, which would harm our business and financial condition.
Our products may be expensive to manufacture, and they may not be profitable if we are unable to control the costs to manufacture them.
Our products may be significantly more expensive to manufacture than we expect or than other therapeutic products currently on the market today. We hope to substantially reduce manufacturing costs through process improvements, development of new methods, increases in manufacturing scale and outsourcing to experienced manufacturers. If we are not able to make these, or other improvements, and depending on the pricing of the product, our profit margins may be significantly less than that of other therapeutic products on the market today. In addition, we may not be able to charge a high enough price for any product we develop, even if they are safe and effective, to make a profit. If we are unable to realize significant profits from our potential product candidates, our business would be materially harmed.
| 23 |
The interruption of the flow of raw materials could disrupt the supply chain and our ability to manufacture our products.
We depend on our outsourced manufacturers to source raw materials for our products. Any shortages of raw materials would affect our ability to timely deliver our products and could significantly harm our business, and results of operations. Other events that could also cause disruptions to our supply chain include the imposition of additional duties, tariffs and other charges or quotas on imports and exports, natural disasters, severe weather, political instability, war, such as the Russia-Ukraine conflict or the Israel-Hamas war, terrorist attacks, and social unrest and economic instability in the regions in which our raw material suppliers are located, or through which regions they travel.
We currently lack manufacturing capabilities to produce our therapeutic product candidates at commercial-scale quantities and do not have an alternate manufacturing supply, which could negatively impact our ability to meet any future demand for the product.
We expect that we would need to significantly expand our manufacturing capabilities to meet potential demand for our therapeutic product candidates, if approved. Such expansion would require additional regulatory approvals. Even if we increase our manufacturing capabilities, it is possible that we may still lack sufficient capacity to meet demand.
We do not currently have any alternate supply for our products. If the facilities where our products are currently being manufactured or equipment were significantly damaged or destroyed, or if there were other disruptions, delays or difficulties affecting manufacturing capacity or availability of drug supply, including, but not limited to, if such facilities are deemed not in compliance with current Good Manufacturing Practice (“GMP”), requirements, future clinical studies and commercial production for our products would likely be significantly disrupted and delayed. It would be both time-consuming and expensive to replace this capacity with third parties, particularly since any new facility would need to comply with the regulatory requirements.
Ultimately, if we are unable to supply our products to meet commercial demand, whether because of processing constraints or other disruptions, delays or difficulties that we experience, our production costs could dramatically increase and sales of our products and their long-term commercial prospects could be significantly damaged.
To be successful, our proposed products must be accepted by the healthcare community, which can be very slow to adopt or unreceptive to new technologies and products.
Our proposed products and those developed by our collaborative partners, if approved for marketing, may not achieve market acceptance since hospitals, physicians, patients or the medical community in general may decide not to accept and use these products. The products that we are attempting to develop represent substantial departures from established treatment methods and will compete with a number of more conventional therapies manufactured and marketed by major pharmaceutical companies. The degree of market acceptance of any of our developed products will depend on a number of factors, including:
| ● | our establishment and demonstration to the medical community of the clinical efficacy and safety of our proposed products; |
| ● | our ability to create products that are superior to alternatives currently on the market; |
| ● | our ability to establish in the medical community the potential advantage of our treatments over alternative treatment methods; and |
| ● | reimbursement policies of government and third-party payers. |
If the healthcare community does not accept our products for any of these reasons, or for any other reason, our business would be materially harmed.
Our business is based on novel technologies that are inherently expensive and risky and may not be understood by or accepted in the marketplace, which could adversely affect our future value.
The clinical development, commercialization and marketing of immuno-oncology therapies are at an early-stage, substantially research-oriented, and financially speculative. To date, very few companies have been successful in their efforts to develop and commercialize an immuno-oncology therapeutic product. In general, such products may be susceptible to various risks, including undesirable and unintended side effects, unintended immune system responses, inadequate therapeutic efficacy, or other characteristics that may prevent or limit their approval or commercial use. Furthermore, the number of people who may use such therapies is difficult to forecast with accuracy. Our future success is dependent on the establishment of a significant market for such therapies and our ability to capture a share of this market with our product candidates.
Our development efforts with our therapeutic product candidates are susceptible to the same risks of failure inherent in the development and commercialization of therapeutic products based on new technologies. The novel nature of immuno-oncology therapeutics creates significant challenges in the areas of product development and optimization, manufacturing, government regulation, third-party reimbursement and market acceptance. For example, the FDA has relatively limited experience regulating such therapies, and there are few approved treatments using such therapy.
| 24 |
Our competition includes fully integrated biotechnology and pharmaceutical companies that have significant advantages over us.
The market for therapeutic immuno-oncology products is highly competitive. We expect that our most significant competitors will be fully integrated and more established pharmaceutical and biotechnology companies or institutions, including major multinational pharmaceutical companies, biotechnology companies and universities and other research institutions. These companies are developing similar products, and they have significantly greater capital resources and research and development, manufacturing, testing, regulatory compliance, and marketing capabilities. Many of these potential competitors may be further along in the process of product development and also operate large, company-funded research and development programs. As a result, our competitors may develop more competitive or affordable products, or achieve earlier patent protection or product commercialization than we are able to achieve. Competitive products may render any products or product candidates that we develop obsolete.
Many of our competitors have substantially greater financial, technical and other resources than we do, such as larger research and development staff and experienced marketing and manufacturing organizations. Additional mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in certain of our competitors. As a result, these companies may be able to obtain regulatory approval more rapidly than we can and may be more effective in selling and marketing their products. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or licensing drug products that are more effective or less costly to produce or purchase on the market than any product candidate we are currently developing or that we may seek to develop in the future. If approved, our product candidates will face competition from commercially available drugs as well as drugs that are in the development pipelines of our competitors.
Established pharmaceutical companies may invest heavily to accelerate discovery and development of or in-license novel compounds that could make our product candidates less competitive. In addition, any new product that competes with an approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and safety in order to overcome price competition and to be commercially successful. Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA, EMA or other regulatory approval, or discovering, developing and commercializing medicines before we do, which would have a material adverse impact on our business and ability to achieve profitability from future sales of our approved product candidates, if any.
If competitors develop and market products that are more effective, safer or less expensive than our product candidates or offer other advantages, our commercial prospects will be limited.
Our therapeutic immuno-oncology (“IO”) development programs face, and will continue to face, intense competition from pharmaceutical, biopharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies engaged in drug discovery activities or funding, both in the United States and abroad. Some of these competitors are pursuing the development of drugs and other therapies that target the same diseases and conditions that we are targeting with our product candidates. According to recent (early 2026) industry research form Global Data and Research and Markets, there are approximately 9,000 industry-sponsored clinical trials for immuno-oncology with more than 1,200 drugs in development. Phase 2 trials constitute the majority of the IO pipeline at approximately 48%, followed by early-stage molecules in Phase 1/2 and Phase 1. Late-stage development remains highly competitive, with over 600 clinical trials ongoing in Phase 3. There are currently over 85 marketed immuno-oncology agents globally. While checkpoint modulators (specifically PD-1/PD-L1 inhibitors) remain the dominant category by revenue, the category of cancer vaccines has seen a resurgence due to breakthroughs in mRNA technology, with over 120 RNA-based vaccine trials currently active. The indications with the most marketed IO agents in the United States remain non-small cell lung cancer and metastatic melanoma, though competition has intensified in triple-negative breast cancer and renal cell carcinoma. The global market value of the IO sector—encompassing bispecific antibodies, cancer vaccines, checkpoint modulators, cell therapies, and oncolytic viruses—has reached an estimated $136 billion in 2025 and is projected to exceed $151 billion by the end of 2026, representing a monumental shift from the $29 billion valuation reported in 2019. The global market for solid tumor cancers accounts is estimated to be $362 billion (according to Data Bridge Market Research).
As a general matter, we also face competition from many companies that are researching and developing cell therapies. Many of these companies have financial and other resources substantially greater than ours. In addition, many of these competitors have significantly greater experience in testing pharmaceutical and other therapeutic products, obtaining FDA and other regulatory approvals, and marketing and selling. If we obtain regulatory approval for any of our product candidates, we also will be competing with respect to manufacturing efficiency and marketing capabilities, areas in which we have limited or no commercial-scale experience. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources’ being concentrated by our competitors. Competition may increase further as a result of advances made in the commercial applicability of our technologies and greater availability of capital for investment in these fields.
| 25 |
If we are unable to keep up with rapid technological changes in our field or compete effectively, we will be unable to operate profitably.
We are engaged in activities in the biotechnology field, which is characterized by extensive research efforts and rapid technological progress. If we fail to anticipate or respond adequately to technological developments, our ability to operate profitably could suffer. Research and discoveries by other biotechnology, pharmaceutical or other companies may render our technologies or potential products or services uneconomical or result in products superior to those we develop. Similarly, any technologies, products or services we develop may not be preferred to any existing or newly developed technologies, products or services.
We may not be able to obtain third-party patient reimbursement or favorable product pricing, which would reduce our ability to operate profitably.
Our ability to successfully commercialize certain of our proposed products in the human therapeutic field may depend to a significant degree on patient reimbursement of the costs of such products and related treatments at acceptable levels from government authorities, private health insurers and other organizations, such as health maintenance organizations. Reimbursement in the United States or foreign countries may not be available for any products we may develop, and, if available, may be decreased in the future. Also, reimbursement amounts may reduce the demand for, or the price of, our products with a consequent harm to our business. We cannot predict what additional regulation or legislation relating to the healthcare industry or third-party coverage and reimbursement may be enacted in the future or what effect such regulation or legislation may have on our business. If additional regulations are overly onerous or expensive, or if healthcare-related legislation makes our business more expensive or burdensome than originally anticipated, we may be forced to significantly downsize our business plans or completely abandon our business model.
We may be subject to litigation that will be costly to defend or pursue and uncertain in its outcome.
Our business may bring us into conflict with our licensees, licensors or others with whom we have contractual or other business relationships, or with our competitors or others whose interests differ from ours. If we are unable to resolve those conflicts on terms that are satisfactory to all parties, we may become involved in litigation brought by or against us. That litigation is likely to be expensive and may require a significant amount of management’s time and attention, at the expense of other aspects of our business. The outcome of litigation is always uncertain, and in some cases could include judgments against us that require us to pay damages, enjoin us from certain activities, or otherwise affect our legal or contractual rights, which could have a significant adverse effect on our business.
We are exposed to the risk of liability claims, for which we may not have adequate insurance.
Since we participate in the pharmaceutical industry, we may be subject to liability claims by employees, customers, end users and third parties. We intend to obtain proper insurance, however, there can be no assurance that any liability insurance we purchase will be adequate to cover claims asserted against us or that we will be able to maintain such insurance in the future. We intend to adopt prudent risk-management programs to reduce these risks and potential liabilities, however, we have not taken any steps to create these programs and have no estimate as to the cost or time required to do so and there can be no assurance that such programs, if and when adopted, will fully protect us. We may not be able to put risk management programs in place, or obtain insurance, if we are unable to retain the necessary expertise and/or are unsuccessful in raising necessary capital in the future. Our failure to obtain appropriate insurance, or to adopt and implement effective risk-management programs, as well as any adverse rulings in any legal matters, proceedings and other matters could have a material adverse effect on our business.
Preclinical and clinical trials are conducted during the development of potential products and other treatments to determine their safety and efficacy for use by humans. Notwithstanding these efforts, when our treatments are introduced into the marketplace, unanticipated side effects may become evident. Manufacturing, marketing, selling and testing our product candidates under development or to be acquired or licensed, entails a risk of product liability claims. We could be subject to product liability claims if our product candidates, processes, or products under development fail to perform as intended. Even unsuccessful claims could result in the expenditure of funds in litigation and the diversion of management time and resources, and could damage our reputation and impair the marketability of our product candidates and processes. While we plan to maintain liability insurance for product liability claims, we may not be able to obtain or maintain such insurance at a commercially reasonable cost. If a successful claim were made against us, and we lacked insurance or the amount of insurance were inadequate to cover the costs of defending against or paying such a claim or the damages payable by us, we would experience a material adverse effect on our business, financial condition and results of operations.
| 26 |
We could be subject to product liability lawsuits based on the use of our product candidates in clinical testing or, if obtained, following marketing approval and commercialization. If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to cease clinical testing or limit commercialization of our product candidates.
We could be subject to product liability lawsuits if any product candidate we develop allegedly causes injury or is found to be otherwise unsuitable for human use during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates, if approved. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our product candidates; |
| ● | withdrawal of clinical trial participants; |
| ● | initiation of investigations by regulators; |
| ● | costs to defend the related litigation; |
| ● | a diversion of management’s time and our resources; |
| ● | substantial monetary awards to trial participants or patients; |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| ● | loss of revenues from product sales; and |
| ● | the inability to commercialize our product candidates. |
Our inability to retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the clinical testing and commercialization of products we develop. We may wish to obtain additional such insurance covering studies or trials in other countries should we seek to expand those clinical trials or commence new clinical trials in other jurisdictions or increase the number of patients in any clinical trials we may pursue. We also may determine that additional types and amounts of coverage would be desirable at later stages of clinical development of our product candidates or upon commencing commercialization of any product candidate that obtains required approvals. However, we may not be able to obtain any such additional insurance coverage when needed on acceptable terms or at all. If we do not obtain or retain sufficient product liability insurance, we could be responsible for some or all of the financial costs associated with a product liability claim relating to our preclinical and clinical development activities, in the event that any such claim results in a court judgment or settlement in an amount or of a type that is not covered, in whole or in part, by any insurance policies we may have or that is in excess of the limits of our insurance coverage. We may not have, or be able to obtain, sufficient capital to pay any such amounts that may not be covered by our insurance policies.
We rely on third parties to conduct preclinical and clinical trials of our product candidates. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
We rely, and expect to continue to rely, upon third-party clinical research organizations (“CROs”) to execute our preclinical and clinical trials and to monitor and manage data produced by and relating to those trials. However, we may not be able to establish arrangements with CROs when needed or on terms that are acceptable to us, or at all, which could negatively affect our development efforts with respect to our drug product candidates and materially harm our business, operations and prospects.
We will have only limited control over the activities of the CROs we will engage to conduct our clinical trials. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on any of the CROs does not relieve us of our regulatory responsibilities. Based on our present expectations, we, our CROs and our clinical trial sites are required to comply with good clinical practices (“GCPs”), for all our product candidates in clinical development. Regulatory authorities enforce GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs fail to comply with applicable GCPs, the clinical data generated in the applicable trial may be deemed unreliable and the FDA, EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving a product candidate for marketing, which we may not have sufficient cash or other resources to support and which would delay our ability to generate revenue from any sales of such product candidate. In addition, our clinical trials are required to be conducted with product produced in compliance with current good manufacturing practice requirements, or cGMP. Our CROs’ failure to comply with those regulations may require us to repeat clinical trials, which would also require significant cash expenditures and delay the regulatory approval process.
| 27 |
Agreements governing relationships with CROs generally provide those CROs with certain rights to terminate a clinical trial under specified circumstances. If a CRO that we have engaged terminates its relationship with us during the performance of a clinical trial, we would be forced to seek an engagement with a substitute CRO, which we may not be able to do on a timely basis or on commercially reasonable terms, if at all, and the applicable trial would experience delays or may not be completed. In addition, our CROs are not our employees, and except for remedies available to us under any agreements we enter with them, we are unable to control whether or not they devote sufficient time and resources to our clinical, nonclinical and preclinical programs. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to a failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for, or successfully commercialize, the affected product candidates. As a result, our operations and the commercial prospects for the effected product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
We contract with third parties for the supply of product candidates for clinical testing and expect to contract with third parties for the manufacturing of our product candidates for large-scale testing and commercial supply. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or products or such quantities at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts.
We anticipate continuing our engagement of third parties to provide our clinical supply as we advance our product candidates into and through clinical development, and we depend on third parties to produce and maintain sufficient quantities of material to supply our clinical trials. If these third parties do not produce and maintain adequate supplies of clinical material, our development efforts could be significantly delayed, or could incur substantially higher costs. We expect in the future to use third parties for the manufacture of our product candidates for clinical testing, as well as for commercial manufacture. We plan to enter into long-term supply agreements with several manufacturers for commercial supplies. We may be unable to reach agreement on satisfactory terms with contract manufacturers to manufacture our product candidates. Additionally, the facilities to manufacture our product candidates must be the subject of a satisfactory inspection before the FDA or other regulatory authorities approve a marketing authorization for the product candidate manufactured at that facility. We will depend on these third-party manufacturers for compliance with the FDA’s and international regulatory authority requirements for the manufacture of our finished products. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturers for compliance with cGMP. If our manufacturers cannot successfully manufacture material that conforms to our specifications and the FDA and other regulatory authorities’ cGMP requirements, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved, and may subject us to recalls or enforcement action for products already on the market.
If any of our product candidates are approved and contract manufacturers fail to deliver the required commercial quantities of finished product on a timely basis and at commercially reasonable prices, and we are unable to find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volumes and quality and on a timely basis, we would likely be unable to meet demand for our products and could lose potential revenue. It may take several years to establish an alternative source of supply for our product candidates and to have any such new source approved by the FDA or any other relevant regulatory authorities.
We currently have no marketing and sales force. If we are unable to establish effective marketing and sales capabilities or enter into agreements with third parties to market and sell our product candidates, we may not be able to effectively market and sell our product candidates, if approved, or generate product revenues.
We currently do not have a marketing or sales team for the marketing, sales and distribution of any of our product candidates that are able to obtain regulatory approval. To commercialize any product candidates, we must build on a territory-by-territory basis marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. If our product candidates receive regulatory approval, we intend to establish an internal sales and marketing team with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive and time consuming and will require significant attention of our executive officers to manage. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of any of our products that we obtain approval to market. With respect to the commercialization of all or certain of our product candidates, we may choose to collaborate, either globally or on a territory-by-territory basis, with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. If we are unable to enter into such arrangements when needed on acceptable terms or at all, we may not be able to successfully commercialize any of our product candidates that receive regulatory approval or any such commercialization may experience delays or limitations. If we are not successful in commercializing our product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
| 28 |
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and we may incur substantial costs to attempt to recover or reproduce the data. If any disruption or security breach resulted in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and/or the further development of our product candidates could be delayed.
We face risks associated with security breaches or cyber-attacks.
We face risks associated with security breaches or cyber-attacks of our computer systems or those of our third-party representatives, vendors, and service providers. Armed conflicts in the Middle East and between Russia and Ukraine, and tensions with countries such as Iran and North Korea and resulting geopolitical uncertainties also could result in an increase in cyberattacks that could either directly or indirectly impact our operations. Although we have implemented security procedures and controls to address these threats, such as firewalls, encryption, access controls, and employee training programs, cybersecurity threats are dynamic and evolving and our systems may still be vulnerable to theft, loss or misuse of data, including proprietary or confidential information, relating to our business, products, employees, suppliers and customers; disruption due to computer viruses and programming errors; attacks by third parties including destruction of data or demanding ransom to return control of our systems and services; or similar disruptive problems.
Our operations are vulnerable to interruption by natural disasters, power loss, terrorist activity and other events beyond our control, the occurrence of which could materially harm our business.
Businesses located in California have, in the past, been subject to electrical blackouts as a result of a shortage of available electrical power, and any future blackouts could disrupt our operations. We are vulnerable to a major earthquake, wildfire and other natural disasters, and we have not undertaken a systematic analysis of the potential consequences to our business as a result of any such natural disaster and do not have an applicable recovery plan in place. We do not carry any business interruption insurance that would compensate us for actual losses from interruption of our business that may occur, and any losses or damages incurred by us could cause our business to materially suffer.
Epidemic or pandemic outbreaks such as COVID-19 (coronavirus), natural disasters, whether or not caused by climate change, unusual weather conditions, terrorist acts and political events, could disrupt business and result in halting our clinical trials and otherwise adversely affect our financial performance.
The occurrence of one or more natural disasters, such as tornadoes, hurricanes, fires, floods and earthquakes, unusual weather conditions, epidemic outbreaks, terrorist attacks or disruptive political events in certain regions where our operations are located could adversely affect our business. Epidemic or pandemic outbreaks, such as COVID-19 (coronavirus) could impact our management and our ability to conduct clinical trials. This also may affect the market conditions that would limit our ability to raise additional capital. This could have a sustained material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Common Stock
Our common stock may be at risk for delisting from Nasdaq in the future if we do not regain or maintain compliance with Nasdaq’s continued listing requirements. Delisting could adversely affect the liquidity of our common stock and the market price of our common stock could decrease.
Our common stock is currently listed on Nasdaq. Nasdaq has minimum requirements that a company must meet in order to remain listed on Nasdaq, including corporate governance standards and the $1 minimum bid price requirement for continued listing on Nasdaq, as set forth in Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Requirement”).
On November 20, 2025, the Company received a letter (the “Letter”) from the Nasdaq Listing Qualifications Staff (the “Staff”) notifying the Company that its common stock had closed below $1 per share for 30 consecutive business days and, as a result, the Company was not in compliance with the Minimum Bid Price Requirement. The Letter had no immediate effect on the listing of the Company’s common stock which continues to trade on Nasdaq under the symbol “GTBP,” subject to the Company’s compliance with the other Nasdaq listing requirements.
In accordance with Nasdaq Listing Rule 5810(c)(3)(A), the Company was provided a compliance period of 180 calendar days from the date of the Letter, or until May 19, 2026 (the “Compliance Period”), to regain compliance with the Minimum Bid Price requirement. If at any time during the Compliance Period, the closing bid price of the Company’s common stock is at least $1.00 per share for a minimum of ten consecutive business days (unless the Nasdaq staff exercises its discretion to extend this ten business day period pursuant to Nasdaq Listing Rule 5810(c)(3)(H)), Nasdaq will provide the Company written confirmation of compliance with the Minimum Bid Price Requirement, and the matter will be closed.
| 29 |
If the Company does not regain compliance during the Compliance Period, the Company may be eligible for an additional 180-calendar day period to regain compliance with the Minimum Bid Price Requirement, provided that it meets the applicable market value of publicly held shares requirement for continued listing and all other applicable standards for initial listing on Nasdaq (except the Minimum Bid Price Requirement), and notifies Nasdaq of its intent to cure the deficiency by effecting a reverse stock split of its common stock, if necessary. If Nasdaq determines that the Company is not eligible for an additional 180 calendar days compliance period or the Company will not be able to cure the deficiency with the Minimum Bid Price Requirement within the allotted compliance period, the Company’s stock will be subject to delisting.
If our common stock is delisted from Nasdaq, our ability to raise capital through public offerings of our securities and to finance our operations could be adversely affected. We also believe that delisting would likely result in decreased liquidity and/or increased volatility in our common stock and could harm our business and future prospects. In addition, we believe that, if our common stock is delisted, our stockholders would likely find it more difficult to obtain accurate quotations as to the price of our common stock and it may be more difficult for stockholders to buy or sell our common stock at competitive market prices, or at all.
In the future, if we fail to maintain such minimum requirements and a final determination is made by Nasdaq that our common stock must be delisted, the liquidity of our common stock would be adversely affected and the market price of our common stock could decrease. In addition, if delisted, we would no longer be subject to Nasdaq rules, including rules requiring us to have a certain number of independent directors and to meet other corporate governance standards. Our failure to be listed on Nasdaq or another established securities market would have a material adverse effect on the value of your investment in us.
We may issue additional shares of common stock or preferred stock, which would dilute the interest of our stockholders and could cause the price of our common stock to decline.
Pursuant to our Restated Certificate of Incorporation (the “Charter”), our authorized shares consist of 250,000,000 shares of common stock and 15,000,000 shares of preferred stock.
To raise capital, we may sell common stock, preferred stock, convertible securities or other equity securities in one or more transactions, including those contemplated by the Committed Equity Facility (as defined below), at prices and in a manner we determine from time to time. We may sell shares or other securities in any other offering at a price per share that is less than the price per share paid by our current stockholders, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders. Any such issuance:
| ● | may significantly dilute the equity interest of our then-current stockholders; | |
| ● | may subordinate the rights of holders of shares of common stock if one or more classes of preferred stock are created, and such preferred shares are issued, with rights senior to those afforded to our common stock; | |
| ● | could cause a change in control if a substantial number of shares of common stock are issued, which may affect, among other things, our ability to use our net operating loss carryforwards, if any, and could result in the resignation or removal of our present officers and directors; and | |
| ● | may adversely affect the prevailing market price for our common stock. |
On May 14, 2025, the Company entered into the Common Shares Purchase Agreement with Bristol and 5NL, relating to the Committed Equity Facility, whereby we have the right from time to time at our option to sell to the Facility Investors up to $20 million of our common stock subject to certain conditions and limitations set forth in the Common Shares Purchase Agreement. Sales of the shares of common stock to the Facility Investors under the Common Shares Purchase Agreement, and the timing of any sales, are determined by the Company from time to time in its sole discretion and depend on a variety of factors, including, among other things, market conditions, the trading price of the common stock and determinations by the Company regarding the use of proceeds of such shares of common stock. The net proceeds from any sales under the Common Shares Purchase Agreement will depend on the frequency with, and prices at, which the shares of common stock are sold to the Facility Investors. The purchase price of the shares of common stock that the Company elects to sell to the Facility Investors pursuant to the Common Shares Purchase Agreement are equal to 93% of the volume weighted average price of the shares of Common Stock during the applicable purchase date on which the Company has timely delivered written notice to the Facility Investors directing it to purchase shares of common stock under the Common Shares Purchase Agreement.
We may not have sufficient authorized shares to issue in connection with a future capital raise without stockholder approval. No assurance can be given that our shareholders will approve an increase in authorized shares, that may result in our inability to raise adequate funds to execute our strategy.
| 30 |
There has been a limited public market for our common stock, and we do not know whether one will develop to provide adequate liquidity. Furthermore, the trading price for our common stock, should an active trading market develop, may be volatile and could be subject to wide fluctuations in per-share price.
Our common stock is now listed on the Nasdaq Capital Market under the trading symbol “GTBP”; historically, however, there has been a limited public market for our common stock. We cannot assure you that an active trading market for our common stock will develop or be sustained. The liquidity of any market for the shares of our common stock will depend on a number of factors, including:
| ● | the number of stockholders; |
| ● | our operating performance and financial condition; |
| ● | the market for similar securities; |
| ● | the extent of coverage of us by securities or industry analysts; and |
| ● | the interest of securities dealers in making a market in the shares of our common stock. |
Even if an active trading market develops, the market price for our common stock may be highly volatile and could be subject to wide fluctuations. In addition, the price of shares of our common stock could decline significantly if our future operating results fail to meet or exceed the expectations of market analysts and investors and actual or anticipated variations in our quarterly operating results could negatively affect our share price.
The volatility of the price of our common stock may also be impacted by the risks discussed under this “Risk Factors” section, in addition to other factors, including:
| ● | developments in the financial markets and worldwide or regional economies; |
| ● | announcements of innovations or new products or services by us or our competitors; |
| ● | announcements by the government relating to regulations that govern our industry; |
| ● | significant sales of our common stock or other securities in the open market; |
| ● | variations in interest rates; |
| ● | changes in the market valuations of other comparable companies; and |
| ● | changes in accounting principles. |
Our outstanding warrants and options may affect the market price of our common stock.
As of December 31, 2025, we had 25,534,173 shares of common stock outstanding and issued and had outstanding warrants for the purchase of up to 50,532,927 additional shares of common stock at a weighted average exercise price of $1.74 per share, 16,344,159 of which are exercisable (subject to certain beneficial ownership limitations). In addition, we had outstanding options for the purchase of up to 597,550 additional shares of common stock at a weighted average exercise price of $4.23 per share, 435,883 of which are exercisable. The amount of common stock reserved for issuance may have an adverse impact on our ability to raise capital and may affect the price and liquidity of our common stock in the public market. In addition, the issuance of these shares of common stock will have a dilutive effect on current stockholders’ ownership.
Because our common stock may be deemed a low-priced “penny” stock, an investment in our common stock should be considered high-risk and subject to marketability restrictions.
Historically, the trading price of our common stock has been $5.00 per share or lower, and deemed a penny stock, as defined in Rule 3a51-1 under the Exchange Act, and subject to the penny stock rules of the Exchange Act specified in rules 15g-1 through 15g-100. Those rules require broker-dealers, before effecting transactions in any penny stock, to:
| ● | deliver to the customer, and obtain a written receipt for, a disclosure document; |
| 31 |
| ● | disclose certain price information about the stock; |
| ● | disclose the amount of compensation received by the broker-dealer or any associated person of the broker-dealer; |
| ● | send monthly statements to customers with market and price information about the penny stock; and |
| ● | in some circumstances, approve the purchaser’s account under certain standards and deliver written statements to the customer with information specified in the rules. |
Consequently, the penny stock rules may restrict the ability or willingness of broker-dealers to sell the common stock and may affect the ability of holders to sell their common stock in the secondary market and the price at which such holders can sell any such securities. These additional procedures could also limit our ability to raise additional capital in the future.
Financial Industry Regulatory Authority (“FINRA”) sales practice requirements may also limit a stockholder’s ability to buy and sell our common stock, which could depress the price of our common stock.
In addition to the “penny stock” rules described above, FINRA has adopted rules that require a broker-dealer to have reasonable grounds for believing that the investment is suitable for that customer before recommending an investment to a customer. Prior to recommending speculative low-priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low-priced securities will not be suitable for at least some customers. Thus, the FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our common stock, which may limit your ability to buy and sell our shares of common stock, have an adverse effect on the market for our shares of common stock, and thereby depress our price per share of common stock.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our stock, our stock price and trading volume could decline.
The trading market for our common stock may be influenced by the research and reports that industry or securities analysts publish about us or our business. We currently have research coverage by only one securities analyst, and we may never obtain research coverage by additional analysts. If no or few securities or industry analysts commence coverage of us, the trading price for our common stock may be negatively affected. In the event that we receive additional securities or industry analyst coverage, if any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or our stock performance, or if our operating results fail to meet the expectations of analysts, our stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
We are subject to state and federal securities laws, the terms of the Charter, Amended and Restated Bylaws (the “Bylaws”) and the terms of complex contracts; a failure to comply with current and future laws, regulations or standards, or the terms of our Charter, Bylaws or contractual arrangements, could have an adverse effect on our business.
We are subject to state and federal securities laws, including the information and reporting requirements of the Securities Exchange Act of 1934, as amended, and other federal securities laws, as well as the provisions of the Delaware General Corporation Law. Failure or noncompliance with such laws, and any regulations or standards could have an adverse effect on our business. In addition, we operate pursuant to our Charter, Bylaws and complex contractual arrangements, including any amendments or waivers thereunder. Our failure to comply with the terms of our Charter, Bylaws, or any such contractual arrangements, or if any amendments or waivers thereunder are deemed unenforceable, we could be exposed to claims or other demands that could divert management’s attention from other business concerns and could have an adverse effect on business, financial condition, and results of operation.
Anti-takeover provisions may limit the ability of another party to acquire us, which could cause our stock price to decline.
Delaware law and our Charter, our Bylaws and other governing documents contain provisions that could discourage, delay or prevent a third party from acquiring us, even if doing so may be beneficial to our stockholders, which could cause our stock price to decline. In addition, these provisions could limit the price investors would be willing to pay in the future for shares of our common stock.
We do not currently or for the foreseeable future intend to pay dividends on our common stock.
We have never declared or paid any cash dividends on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. As a result, any return on your investment in our common stock will be limited to the appreciation in the price of our common stock, if any.
| 32 |
We are subject to ongoing regulatory burdens resulting from our public listing.
We continually work with our legal, accounting and financial advisors to identify those areas in which changes should be made to our financial management control systems to manage our obligations as a public company listed on Nasdaq. These areas include corporate governance, corporate controls, disclosure controls and procedures and financial reporting and accounting systems. However, these and other measures that we might take may not be sufficient to allow us to satisfy our obligations as a public company listed on Nasdaq on a timely basis. In addition, compliance with reporting and other requirements applicable to public companies listed on Nasdaq creates additional costs for us and requires the time and attention of management. The additional costs that we incur, the timing of such costs and the impact that management’s attention to these matters may adversely affect our business and operating results.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, stockholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our common stock.
We produce our financial statements in accordance with accounting principles generally accepted in the United States (“GAAP”). Effective internal control over financial reporting is necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could cause us to fail to meet our reporting obligations. Ineffective internal control could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
As defined in Regulation 12b-2 under the Securities Exchange Act of 1934, or the Exchange Act, a “material weakness” is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented, or detected on a timely basis. We have taken measures to mitigate potential issues and have implemented a functional system of internal controls over financial reporting. However, such controls may become inadequate due to changes in conditions, or the degree of compliance with such policies or procedures may deteriorate, which could result in the discovery of material weaknesses and deficiencies.
As a public company, we are required to document and test our internal control procedures in order to satisfy the requirements of Section 404 of the Sarbanes-Oxley Act of 2002 (“Section 404”). The process of determining whether our existing internal control over financial reporting is compliant with Section 404, and sufficiently effective requires the investment of substantial time and resources, including by certain members of our senior management. At December 31, 2025, we identified a material weakness in our internal control over financial reporting and concluded that our internal control over financial reporting was not effective. The material weakness related to controls over the accounting for complex financial transactions, specifically the application of ASC 480, Distinguishing Liabilities from Equity, to certain stock purchase rights (the “Greenshoe Rights”), and resulted in the restatement of our previously issued interim financial statements for the quarters ended June 30, 2025 and September 30, 2025.
Although we have implemented remedial measures, we cannot assure you that these efforts will be sufficient to remediate the material weakness or prevent future material misstatements. If we fail to maintain effective internal control over financial reporting, our ability to report accurate financial results on a timely basis could be adversely affected, which could negatively impact investor confidence and the market price of our common stock.
We are also required, pursuant to Section 404, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. However, for as long as we are a “smaller reporting company,” our independent registered public accounting firm will not be required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404. While we could be a smaller reporting company for an indefinite amount of time, and thus relieved of the above-mentioned attestation requirement, an independent assessment of the effectiveness of our internal control over financial reporting could detect problems that our management’s assessment might not. Such undetected material weaknesses in our internal control over financial reporting could lead to financial statement restatements and require us to incur the expense of remediation.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 1C. CYBERSECURITY
| ● | Governance
and Oversight: |
| 33 |
| ● | Risk
Assessment and Management: | |
| ● | Information Security Policies and Procedures: We have established information security policies and procedures that govern the use, protection, and handling of sensitive information. These policies cover areas such as data encryption, access controls, password management, incident response, and employee training. | |
| ● | Network and Infrastructure Security: We employ a range of technical measures to secure our network and infrastructure, including firewalls, intrusion detection and prevention systems, and vulnerability assessments. We also use encryption to protect data both in transit and at rest. | |
| ● | Employee Training and Awareness: We provide cybersecurity training to our employee and consultants to raise awareness of the latest threats and best practices for protecting sensitive information. This includes training on how to recognize phishing attempts, handling secure data, and reporting security incidents. | |
| ● | Third-Party
Risk Management: | |
| ● | Compliance and Reporting: We comply with all applicable laws and regulations related to cybersecurity, including data protection and privacy laws. |
ITEM 2. PROPERTIES
Effective as of July 1, 2024, the Company became a fully remote company. We operate in a virtual environment and do not have a physical office space or headquarters.
ITEM 3. LEGAL PROCEEDINGS
Ohri Matter
On July 22, 2024, the Company filed an AAA Arbitration Demand against Manu Ohri, its former Chief Financial Officer. In the demand, the Company asserts claims against Mr. Ohri for breach of his fiduciary duties and breach of contract and seeks a declaratory judgment providing that the Company may characterize Mr. Ohri’s termination as “for cause” under his employment agreement, and that the Company may revoke the separation agreement entered into between the Company and Mr. Ohri prior to the Company learning of Mr. Ohri’s breaches. In addition to the declaratory judgment, the Company seeks damages arising from Mr. Ohri’s violations, and attorneys’ fees and any forum and arbitration fees. On September 3, 2024, Mr. Ohri filed both a general denial of the Company’s claims against him and counterclaims for breach of his employment agreement and separation agreement. The final hearing date, originally scheduled for June 10, 2025, has been postponed indefinitely in order to allow the parties to mediate the dispute. Mediation is scheduled for March 31, 2026. Should mediation be unsuccessful, the case will proceed to a final hearing in October 2026. At this stage in the proceedings the Company is not able to determine the probability of the outcome of this matter or a range of reasonably expected losses, if any. The Company believes it has recorded an appropriate accrual for the matter.
| 34 |
TWF Global Matter
On May 24, 2023, TWF Global, LLC (“TWF”) filed a Complaint in the California Superior Court for the County of Los Angeles naming the Company as defendant. The complaint alleges that TWF is the holder of two Convertible Promissory Notes (“Notes”) and that the Company did not deliver shares of common stock due on conversion in February 2021. TWF was seeking per diem liquidated damages based on the terms of alleged Notes. On July 14, 2023, the Company filed a motion to dismiss for improper forum because the terms of the Notes, as alleged, require disputes to be filed in New York state and federal courts. TWF voluntarily dismissed its complaint before the California Superior Court of Los Angeles without prejudice. The Company subsequently filed a summons and complaint for interpleader against TWF and Z-One, LLC before the Supreme Court of the State of New York County of New York, asking the Supreme Court to determine if the Company’s shares of common stock should be registered to TWF or Z-One LLC, as both of these entities have made conflicting demands for the shares. On February 5, 2024, the Company filed a motion for entry of default against TWF, seeking an order directing the Company to register the shares of common stock in the name of Z-One, LLC and that the Company be released from all associated liability and claims. The Court denied the motion without prejudice and agreed to reconsider the motion without further briefing upon the filing of a supplemental party affidavit. On May 9, 2024, Z-One, LLC filed a motion for summary judgement seeking dismissal of the action, representing that Z-One, LLC and TWF have settled their dispute over the entitlement to the Company’s shares of common stock and there is no remaining dispute before the Court. On May 21, 2024, the Company filed a supplemental affidavit in support of its motion for entry of default. On November 14, 2024, the Court held a hearing on the parties’ motions, at which the Court found that the motion for entry of default was mooted by the settlement agreement between Z-One, LLC and TWF. The Court ordered that the case be dismissed. On February 17, 2025, Z-One, LLC filed a Summons with Notice in the Supreme Court of the State of New York, County of New York. The Company then filed a demand that Z-One, LLC serve a complaint, and on June 25, 2025, Z-One, LLC filed a Complaint alleging that it is the holder, either originally or by assignment, of a Convertible Note in the principal amount of $150,000, that the Company breached the Convertible Note by failing to deliver conversion shares to Z-One, LLC, and that the Company owes it damages in excess of $500,000. On August 26, 2025, the Company filed a motion to dismiss the Complaint in its entirety for lack of standing and failure to state a cause of action. On February 27, 2026 the court issued a Decision and Order granting the Company’s motion and dismissing the claims asserted in the Complaint in their entirety and with prejudice.
Silberfein, DiPietro, and Werthman Trust Matters
On July 8, 2025, Coby Silberfein filed a summons with notice in the State of New York Supreme Court for the County of New York naming the Company as defendant seeking damages for breach of a securities purchase agreement and convertible note in the principal amount of $100,000. On July 8, 2025, Justin DiPietro filed a summons with notice in the State of New York Supreme Court for the County of New York naming the Company as defendant seeking damages for breach of a securities purchase agreement and convertible note in the principal amount of $100,000. On July 9, 2025, Phillip Werthman Trust filed a summons with notice in the State of New York Supreme Court for the County of New York naming the Company as defendant seeking damages for breach of a securities purchase agreement and convertible note in the principal amount of $100,000. The three summons with notice are identical and allege that the plaintiffs are holders of convertible notes and that the Company breached the convertible notes by failing to deliver shares of common stock due on conversion in in 2021. Plaintiffs are seeking specific performance and damages. On August 12, 2025, the Company filed demands that the plaintiffs serve complaints. On September 2, 2025, each plaintiff served a Complaint similar in substance to the summons, except that Plaintiff Silberfein now alleges breach of a convertible note with a principal amount of $150,000, rather than $100,000. Each plaintiff alleges that the Company breached a convertible note by failing to deliver conversion shares to the plaintiff holder, and that the Company owes damages in excess of $500,000. The Company intends to seek dismissal of the complaints. The Company intends to continue to vigorously defend against these claims.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
| 35 |
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Our common stock is traded on the Nasdaq Capital Market under the trading symbol “GTBP.”
Stockholders
As of February 23, 2026, there were 39 stockholders of record, which does not include stockholders who hold their shares in “street name.”
Dividends
We have not paid any dividends on our common stock to date and do not anticipate that we will pay dividends in the foreseeable future. Any payment of cash dividends on our common stock in the future will depend upon the amount of funds legally available, our earnings, if any, our financial condition, our anticipated capital requirements and other factors that the Board of Directors may think are relevant. In addition, our ability to pay dividends may be limited by covenants of any existing and future outstanding indebtedness or agreements we incur or enter into. We do not expect to pay any dividends on our common stock in the foreseeable future.
Equity Compensation Plan Information
Information relating to compensation plans under which our equity securities are authorized for issuance is set forth in Item 12 of this report under “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.”
Recent Issuances of Unregistered Securities
There were no unregistered sales of equity securities sold during the period covered by this Annual Report on Form 10-K that were not previously included in a Quarterly Report on Form 10-Q or in a Current Report on Form 8-K.
Issuer Purchases of Equity Securities
We did not repurchase any shares during the fourth quarter of the fiscal year covered by this Annual Report on Form 10-K.
| 36 |
ITEM 6. [RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Some of the statements in this Annual Report on Form 10-K are “forward-looking statements” within the meaning of the safe harbor from liability established by the Private Securities Litigation Reform Act of 1995. Forward-looking statements include statements regarding our current beliefs, goals and expectations about matters such as our expected financial position and operating results, our business strategy and our financing plans. The forward-looking statements in this report are not based on historical facts, but rather reflect the current expectations of our management concerning future results and events. The forward-looking statements generally can be identified by the use of terms such as “believe,” “expect,” “anticipate,” “intend,” “plan,” “foresee,” “may,” “guidance,” “estimate,” “potential,” “outlook,” “target,” “forecast,” “likely” or other similar words or phrases. Similarly, statements that describe our objectives, plans or goals are, or may be, forward-looking statements. Forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be different from any future results, performance and achievements expressed or implied by these statements. We cannot guarantee that our forward-looking statements will turn out to be correct or that our beliefs and goals will not change. Our actual results could be very different from and worse than our expectations for various reasons. You should carefully review all information, including the discussion of risk factors under “Part I. Item 1A: Risk Factors” and elsewhere in this annual report. Any forward-looking statements in this Form 10-K are made only as of the date hereof and, except as may be required by law, we do not have any obligation to publicly update any forward-looking statements contained in this Form 10-K to reflect subsequent events or circumstances.
Overview
We are a clinical stage biopharmaceutical company focused on the development and commercialization of novel immuno-oncology products based off our proprietary Tri-specific Killer Engager (TriKE®) technology platform. Our TriKE® platform generates proprietary therapeutics designed to harness and enhance the cancer killing abilities of a patient’s own natural killer cells, or NK cells. Once bound to an NK cell, our moieties are designed to enhance the NK cell, and precisely direct it to one or more specifically-targeted proteins expressed on a specific type of cancer cell or virus infected cell, ultimately resulting in the targeted cell’s death. TriKE® is composed of recombinant fusion proteins and interleukin 15 (IL-15), can be designed to target any number of tumor antigens on hematologic malignancies, sarcomas or solid tumors and do not require patient-specific customization.
Critical Accounting Policies
The preparation of our financial statements in conformity with accounting principles generally accepted in the United States (“GAAP”) requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue and expenses, and related disclosure of contingent assets and liabilities. When making these estimates and assumptions, we consider our historical experience, our knowledge of economic and market factors and various other factors that we believe to be reasonable under the circumstances. Actual results may differ under different estimates and assumptions. The accounting estimates and assumptions discussed in this section are those that we consider to be the most critical to gain an understanding of our financial statements because they inherently involve significant judgments and uncertainties.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates include accruals for potential liabilities, assumptions used in deriving the fair value of warrant liabilities, valuation of equity instruments issued for services, and valuation of deferred tax assets. Actual results could differ from those estimates.
Warrant Liability
We evaluate our financial instruments to determine if such instruments are derivatives or contain features that qualify as embedded derivatives in accordance with ASC Topic 815, “Derivatives and Hedging.” For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the statements of operations.
Our use of derivative financial instruments is generally limited to warrants issued by us that do not meet the criteria for equity treatment and are recorded as liabilities. We do not use financial instruments or derivatives for any trading purposes.
Stock-Based Compensation
We periodically issue stock-based compensation to officers, directors, employees, and consultants for services rendered. Such issuances vest and expire according to terms established at the issuance date.
| 37 |
Stock-based payments made to officers, directors, employees, and consultants in exchange for goods and services, including grants of employee stock options, are recognized in the financial statements based on their grant date fair values in accordance with ASC 718, Compensation-Stock Compensation. Stock based payments to officers, directors, employees, and consultants, which are generally time vested, are measured at the grant date fair value and depending on the conditions associated with the vesting of the award, compensation cost is recognized on a straight-line or graded basis over the vesting period. Recognition of compensation expense for non-employees is in the same period and manner as if we had paid cash for the services. The fair value of stock options granted is estimated using the Black-Scholes option-pricing model, which uses certain assumptions related to risk-free interest rates, expected volatility, expected life, and future dividends. The assumptions used in the Black-Scholes option pricing model could materially affect compensation expense recorded in future periods.
Results of Operations
Comparison of the Years Ended December 31, 2025 and 2024
Operating Expenses
| Years Ended December 31, | ||||||||||||||||
| 2025 | 2024 | $ Change | % Change | |||||||||||||
| Operating Expenses: | ||||||||||||||||
| Research and development | $ | 3,472,000 | $ | 5,798,000 | $ | (2,326,000 | ) | (40 | )% | |||||||
| Selling, general and administrative | 8,518,000 | 8,336,000 | 182,000 | 2 | % | |||||||||||
| Stock compensation | 432,000 | 230,000 | 202,000 | 88 | % | |||||||||||
| Total Operating Expenses | $ | 12,422,000 | $ | 14,364,000 | $ | (1,942,000 | ) | (14 | )% | |||||||
Research and Development Expenses
Research and development expenses decreased by approximately $2.3 million for the year ended December 31, 2025, compared to the prior year, primarily due to a decrease in production and materials costs.
Research and development expenses relate to our continued licensing, development and production of our most advanced TriKE® product candidates GTB-3650 and GTB-5550 along with the progression on other promising candidates. In late June 2024, we received clearance from the FDA with respect to our IND Application in relation to our next generation GTB-3650 camelid nanobody product. Study enrollment began in early 2025 and we have advanced into the clinic, enrolling patients, and performing tests for data collection throughout the year. In late January 2026, we received clearance from the FDA with respect to our IND Application in relation to GTB-5550, with a Phase 1 dose escalation basket trial that is expected to initiate mid-2026.
Selling, General, and Administrative Expenses
Selling, general, and administrative expenses remained relatively flat, increasing by only $182,000 for the year ended December 31, 2025, compared to the prior year.
Other Income (Expense)
| Years Ended December 31, | ||||||||||||||||
| 2025 | 2024 | $ Change | % Change | |||||||||||||
| Other Income (Expense): | ||||||||||||||||
| Interest income | $ | 123,000 | $ | 402,000 | $ | (279,000 | ) | (69 | )% | |||||||
| Interest expense | (127,000 | ) | — | (127,000 | ) | 100 | % | |||||||||
| Change in fair value of warrant liability | 241,000 | 800,000 | (559,000 | ) | (70 | )% | ||||||||||
| Loss on initial recognition of Greenshoe Rights liability | (28,736,000 | ) | — | (28,736,000 | ) | 100 | % | |||||||||
| Change in fair value of Greenshoe Rights liability | 11,413,000 | — | 11,413,000 | 100 | % | |||||||||||
| Gain on settlement of vendor payable | 998,000 | — | 998,000 | 100 | % | |||||||||||
| Other | 156,000 | — | 156,000 | 100 | % | |||||||||||
| Total Other Income (Expense) | $ | (15,932,000 | ) | $ | 1,202,000 | $ | (17,134,000 | ) | (1,425 | )% | ||||||
| 38 |
Interest Income
Interest income decreased by $279,000 for the year ended December 31, 2025, compared to the prior year primarily due to lower highly liquid and short-term investment balances.
Interest Expense
Interest expense of $127,000 for the year ended December 31, 2025, consists of straight line amortization of the pre-funded warrants underlying an aggregate of 300,000 shares of common stock issued in connection with the Committed Equity Facility.
Change in Fair Value of Warrant Liability
The change in fair value of warrant liability decreased by $559,000 for the year ended December 31, 2025, compared to the prior year, primarily due to the decline in the Company’s stock price at December 31, 2025, as compared to the prior year.
Loss on Initial Recognition and Change in Fair Value of Greenshoe Rights Liability
The Greenshoe Rights connected to the Series L Preferred Stock issued in the May 2025 equity offering required classification as a liability and marked to market at each reporting date as required under GAAP. The Company recognized other income (expense) for the change in the fair value of the Greenshoe Rights liability at each reporting date. The Greenshoe Rights liability was extinguished and reclassified to equity as of the date of the redemption rights waiver of the Series L 10% Convertible Preferred Stock effective in September 2025 (see Note 10 of the accompanying financial statements).
Gain on Settlement of Vendor Payable
In June 2025, a legal services firm currently engaged by the Company agreed to reduce the Company’s prior year unpaid fees by approximately $1 million..
Other
In May 2025 the Company issued warrants underlying 24,390 shares of common stock exercisable at $2.24 per share with a fair value of approximately $44,000 in exchange for the waiver of a variable rate transaction (“VRT”). The Company classified this transaction as other expense. The Company also recorded other income of $200,000 in June 2025 due to the extinguishment of an accrual of consulting fees recorded in prior years.
Net Loss
| Years Ended December 31, | ||||||||||||||||
| 2025 | 2024 | $ Change | % Change | |||||||||||||
| Net Loss | $ | (28,354,000 | ) | $ | (13,162,000 | ) | $ | (15,192,000 | ) | 115 | % | |||||
Net loss increased approximately $15.2 million for the year ended December 31, 2025, primarily due to the change in fair value of greenshoe rights liability, slightly offset by a decrease in research and development expenses, as described above.
Liquidity and Capital Resources
The accompanying financial statements have been prepared assuming that we will continue as a going concern. We do not have any product candidates approved for sale and have not generated any revenue from our product sales. We have sustained operating losses since inception, and we expect such losses to continue into the foreseeable future. Historically, we have financed our operations through public and private sales of common stock, the issuance of preferred and common stock, the issuance of convertible debt instruments, and strategic collaborations. For the year ended December 31, 2025, we recorded a net loss of approximately $28.4 million and used cash in operations of approximately $12.9 million. These factors raise substantial doubt about our ability to continue as a going concern within one year of the date that the financial statements are issued. In addition, the Company’s independent registered public accounting firm, in its report on the Company’s December 31, 2025 financial statements, raised substantial doubt about the Company’s ability to continue as a going concern. The financial statements do not include any adjustments that might be necessary if the Company is unable to continue as a going concern.
The financial statements do not include any adjustments that might be necessary if we are unable to continue as a going concern. Accordingly, the financial statements have been prepared on a basis that assumes we will continue as a going concern, and which contemplates the realization of assets and satisfaction of liabilities and commitments in the ordinary course of business.
| 39 |
We have evaluated the significance of the uncertainty regarding our financial condition in relation to our ability to meet our obligations, which has raised substantial doubt about our ability to continue as a going concern. While it is very difficult to estimate our future liquidity requirements we will need to obtain additional financing. If we are unable to obtain additional financing, we believe existing cash resources will not be sufficient to enable us to fund the anticipated level of operations through one year from the date the accompanying financial statements are issued. There can be no assurances that we will be able to secure additional financing on acceptable terms. In the event that we do not secure additional financing, we will be forced to delay, reduce, or eliminate some or all of our discretionary spending, which could adversely affect our business prospects, ability to meet long-term liquidity needs and the ability to continue operations.
Cash Flows
| Years Ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Statements of Cash Flow Data: | ||||||||
| Net cash used in operating activities | $ | (12,914,000 | ) | $ | (12,904,000 | ) | ||
| Net cash provided by investing activities | — | 12,893,000 | ||||||
| Net cash provided by financing activities | 15,775,000 | 2,976,000 | ||||||
| Net increase in cash and cash equivalents and restricted cash | 2,861,000 | 2,965,000 | ||||||
| Cash and cash equivalents and restricted cash, beginning of period | 4,044,000 | 1,079,000 | ||||||
| Cash and cash equivalents and restricted cash, end of period | $ | 6,905,000 | $ | 4,044,000 | ||||
Operating Activities
Net cash used in operating activities was approximately $12.9 million for the year ended December 31, 2025, and was primarily due to a net loss of approximately $28.4 million and a decrease in accounts payable and accrued expenses of $2.6 million, offset by the reclassification the Greenshoe Right liability to equity amounting to approximately $17.3 million.
Net cash used in operating activities was approximately $12.9 million for the year ended December 31, 2024, and was primarily due to a net loss of approximately $13.2 million, a decrease in the fair value of warrant liability of $800,000, and partially offset an increase in accounts payable and accrued expenses of approximately $900,000.
Investing Activities
Net cash provided by financing activities for the year ended December 31, 2024, resulted primarily from proceeds from the sale of short-term investments.
Financing Activities
Net cash provided by financing activities was approximately $16 million for the year ended December 31, 2025, and resulted from net proceeds from the issuance of Series L Preferred Stock and warrants, and net proceeds from the exercise of warrants for cash and inducement warrants.
Net cash provided by financing activities approximately $3.0 million for the year ended December 31, 2024, and resulted from net proceeds from issuance of common stock and warrants.
Working Capital (Deficit)
The following table summarizes total current assets, liabilities, and working capital (deficit) for the years ended December 31, 2025 and 2024:
| As of December 31 | ||||||||||||
| 2025 | 2024 | Increase/(Decrease) | ||||||||||
| Current assets | $ | 8,106,000 | $ | 4,232,000 | $ | 3,874,000 | ||||||
| Current liabilities | $ | 2,319,000 | $ | 5,902,000 | $ | (3,583,000 | ) | |||||
| Working capital (deficit) | $ | 5,787,000 | $ | (1,670,000 | ) | $ | 7,457,000 | |||||
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements as of December 31, 2025.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
This company qualifies as a smaller reporting company, as defined in 17 C.F.R. §229.10(f) (1) and is not required to provide information by this Item.
| 40 |
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Stockholders and Board of Directors of GT Biopharma, Inc.
San Francisco, California
Opinion on the Financial Statements
We have audited the accompanying balance sheets of GT Biopharma, Inc. (the “Company”) as of December 31, 2025 and 2024, and the related statements of operations, changes in stockholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2025 and 2024, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, during the year ended December 31, 2025, the Company incurred a net loss of $28.7 million and, used cash in operations of $12.9 million These matters raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1 to the financial statements. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| 41 |
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Accounting for and Valuation of Additional Investment (“Greenshoe”) Rights
As described in Note 5 to the financial statements, the Company issued Series L 10% Convertible Preferred Stock (the “Series L Preferred Stock”) in May 2025 that included additional investment rights (the “Greenshoe Rights”). Due to redemption provisions in the Series L Preferred Stock that could require cash settlement upon events outside the Company’s control, management determined that the Greenshoe Rights met the criteria for liability classification under ASC 480 and were initially recognized at fair value, with subsequent changes in fair value recorded in earnings. In September 2025, the Company executed a waiver eliminating the redemption provisions in the Series L Preferred Stock resulting in remeasurement of the Greenshoe Right and reclassification to equity.
We identified the accounting and valuation of the Greenshoe Rights as a critical audit matter due to the significant auditor judgment required to evaluate the application of ASC 480, the accounting impact and timing of the September 2025 modification, and the fair value measurement of the Greenshoe Rights prior to reclassification.
Our audit procedures related to this matter included, among others:
| ● | We obtained and examined the Securities Purchase Agreement, Certificate of Designation, and waiver agreements to assess redemption provisions and modification terms; | |
| ● | We evaluated the reasonableness of the Company’s technical accounting analysis supporting liability classification prior to the waiver and equity classification thereafter; | |
| ● | We tested the valuation methodology, key assumptions, and mathematical accuracy of the Greenshoe Rights fair value measurement; and | |
| ● | We evaluated the adequacy of the Company’s disclosures related to the financing and related accounting conclusions. |
/s/
PCAOB Firm ID:
March 2, 2026
| 42 |
GT BIOPHARMA, INC.
Balance Sheets
| December 31, 2025 | December 31, 2024 | |||||||
| ASSETS | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Restricted cash | ||||||||
| Deferred offering costs | ||||||||
| Prepaid expenses and other current assets | ||||||||
| Total Current Assets | ||||||||
| TOTAL ASSETS | $ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | $ | ||||||
| Accrued expenses | ||||||||
| Warrant liability | ||||||||
| Total Current Liabilities | ||||||||
| Stockholders’ Equity (Deficit) | ||||||||
| Convertible Preferred stock, par value $, shares authorized: | ||||||||
| Series C - shares issued and outstanding at December 31, 2025 and 2024, respectively | ||||||||
| Series L – and shares issued and outstanding at December 31, 2025 and 2024, respectively | ||||||||
| Common stock, par value $, shares authorized, and shares issued and outstanding as of December 31, 2025 and 2024, respectively | ||||||||
| Additional paid in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total Stockholders’ Equity (Deficit) | ( | ) | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | $ | $ | ||||||
The accompanying notes are an integral part of these financial statements.
| 43 |
GT BIOPHARMA, INC.
Statements of Operations
| For the Year Ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Revenues | $ | $ | ||||||
| Operating Expenses: | ||||||||
| Research and development | ||||||||
| Selling, general and administrative (including $ and $ of stock compensation during the years ended December 31, 2025 and 2024, respectively) | ||||||||
| Loss from Operations | ( | ) | ( | ) | ||||
| Other Income (Expense) | ||||||||
| Interest income | ||||||||
| Interest expense | ( | ) | ||||||
| Loss on initial recognition of Greenshoe Rights liability | ( | ) | ||||||
| Change in fair value of Greenshoe Rights liability | ||||||||
| Change in fair value of warrant liability | ||||||||
| Gain on settlement of vendor payable | ||||||||
| Other | ||||||||
| Total Other Income (Expense), Net | ( | ) | ||||||
| Net Loss | ( | ) | ( | ) | ||||
| Dividend on Series L Preferred Stock | ( |
) | ||||||
| Deemed dividend | ( | ) | ||||||
| Net Loss attributable to common stockholders’ | $ | ( | ) | $ | ( | ) | ||
| Net Loss Per Share - Basic and Diluted | $ | ) | $ | ) | ||||
| Weighted average common shares outstanding - basic and diluted | ||||||||
The accompanying notes are an integral part of these financial statements.
| 44 |
GT BIOPHARMA, INC.
Statements of Stockholders’ Equity (Deficit)
Series C Preferred Shares | Series L Preferred Shares | Common Shares | Additional Paid in | Accumulated | Total Shareholders’ Equity | |||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Capital | Deficit | (Deficit) | ||||||||||||||||||||||||||||
| Balance, December 31, 2023 | $ | $ | $ | $ | $ | ( | ) | $ | ||||||||||||||||||||||||||||
| Common stock and warrants issued for cash | - | - | ||||||||||||||||||||||||||||||||||
| Cancellation of common stock previously issued to prior CFO | - | - | ( | ) | ||||||||||||||||||||||||||||||||
| Issuance of common stock in settlement of vendor payable | - | - | ||||||||||||||||||||||||||||||||||
| Fair value of vested stock options | - | - | - | |||||||||||||||||||||||||||||||||
| Net loss | - | - | - | ( | ) | ( | ) | |||||||||||||||||||||||||||||
| Balance, December 31, 2024 | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||||||||||||||||
| Exercise of warrants for cash and inducement warrants, net | - | - | ||||||||||||||||||||||||||||||||||
| Series L Preferred Stock and warrants issued for cash, net | - | - | ||||||||||||||||||||||||||||||||||
| Conversion of Series L convertible preferred stock into common stock | - | ( | ) | ( | ) | |||||||||||||||||||||||||||||||
| Reclassification of Greenshoe Rights liability to equity | - | - | - | |||||||||||||||||||||||||||||||||
| Issuance of prefunded warrants in settlement of vendor payable | - | - | - | |||||||||||||||||||||||||||||||||
| Issuance and exercise of prefunded warrants for Committed Equity Facility fee | - | - | ||||||||||||||||||||||||||||||||||
| Issuance of common stock and warrants for services | - | - | ||||||||||||||||||||||||||||||||||
| Issuance of warrants for VRT waiver | - | - | - | |||||||||||||||||||||||||||||||||
| Dividend on Series L Preferred Stock | - | - | ( | ) | ( | ) | ||||||||||||||||||||||||||||||
| Fair value of vested stock options | - | - | - | |||||||||||||||||||||||||||||||||
| Net loss | - | - | - | ( | ) | ( | ) | |||||||||||||||||||||||||||||
| Balance, December 31, 2025 | $ | $ | $ | $ | $ | ( | ) | $ | ||||||||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
| 45 |
GT BIOPHARMA, INC.
Statements of Cash Flows
| For the Year Ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock based compensation | ||||||||
| Loss on initial recognition of Greenshoe Rights liability | ||||||||
| Change in fair value of Greenshoe Rights liability | ( | ) | ||||||
| Change in fair value of warrant liability | ( | ) | ( | ) | ||||
| Gain on settlement of debt | ( | ) | ||||||
| Issuance of warrants for VRT waiver | ||||||||
| Gain on extinguishment of debt | ( | ) | ||||||
| Changes in operating assets and liabilities: | ||||||||
| (Increase) Decrease in prepaid expenses and deferred offering costs | ( | ) | ||||||
| Decrease in operating lease right-of-use assets | ||||||||
| Increase (Decrease) in accounts payable and accrued expenses | ( | ) | ||||||
| (Decrease) in operating lease liability | ( | ) | ||||||
| Net Cash Used in Operating Activities | ( | ) | ( | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||
| Sale of short-term investments | ||||||||
| Net Cash Provided by Investing Activities | ||||||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||
| Exercise of warrants and issuance of inducement warrants for cash, net | ||||||||
| Proceeds from issuance of common stock and warrants, net | ||||||||
| Proceeds from issuance of Series L Preferred Stock and warrants, net | ||||||||
| Payment of dividend on Series L Preferred Stock | ( | ) | ||||||
| Net Cash Provided by Financing Activities | ||||||||
| Net Increase in Cash and Cash Equivalents and Restricted Cash | ||||||||
| Cash and Cash Equivalents and Restricted Cash at Beginning of Period | ||||||||
| Cash and Cash Equivalents and Restricted Cash at End of Period | $ | $ | ||||||
| SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION: | ||||||||
| Cash paid during the year for: | ||||||||
| Interest | $ | $ | ||||||
| Income taxes | $ | $ | ||||||
| SUPPLEMENTAL DISCLOSURES OF NON-CASH INVESTING AND FINANCING ACTIVITIES | ||||||||
| Fair value of prefunded warrants issued for Committed Equity Facility fee | $ | $ | ||||||
| Fair value of common stock and warrants issued for services | $ | $ | ||||||
| Fair value of prefunded warrant and common stock to settle vendor payable | $ | $ | ||||||
| Fair value of common stock issued for dividends | $ | $ | ||||||
| Reclassification of Greenshoe Rights liability to equity | $ | 17,232,000 | ||||||
The accompanying notes are an integral part of these financial statements.
| 46 |
GT Biopharma, Inc.
Notes to Financial Statements
For the Years Ended December 31, 2025 and 2024
Note 1 – Organization and Going Concern Analysis
Organization
The corporate predecessor of GT Biopharma, Inc, Diagnostic Data, Inc., was incorporated in the state of California in 1965. Diagnostic Data, Inc. changed its incorporation to the state of Delaware on December 21, 1972 and changed its name to DDI Pharmaceuticals, Inc. on March 11, 1985. On September 7, 1994, DDI Pharmaceuticals, Inc. merged with International BioClinical, Inc. and Bioxytech S.A. and changed its name to OXIS International, Inc. On July 17, 2017, OXIS International, Inc. changed its name to GT Biopharma, Inc.
Throughout this Annual Report on Form 10-K (the “Annual Report”), the terms “GTBP,” “we,” “us,” “our,” “the Company” and “our Company” refer to GT Biopharma, Inc.
The GT Biopharma logo, TriKE®, and other trademarks or service marks of the Company appearing in this Annual Report are the property of the Company. This Annual Report also contains registered marks, trademarks and trade names of other companies. All other trademarks, registered marks and trade names appearing herein are the property of their respective holders.
The Company is a clinical stage biopharmaceutical company focused on the development and commercialization of novel immune-oncology products based on our proprietary Tri-specific Killer Engager (“TriKE®”), and Tetra-specific Killer Engager (“Dual Targeting TriKE®”) platforms. The Company’s TriKE® and Dual Targeting TriKE® platforms generate proprietary therapeutics designed to harness and enhance the cancer killing abilities of a patient’s own natural killer cells (“NK cells”).
Going Concern Analysis
The
accompanying financial statements have been prepared assuming that the Company will continue as a going concern. The Company does not
have any product candidates approved for sale and has not generated any revenue from its product sales. The Company has sustained operating
losses since inception, and expects such losses to continue into the foreseeable future. Historically, the Company has financed its operations
through public and private sales of common stock, issuances of preferred and common stock, issuances of convertible debt instruments, and
strategic collaborations. For the year ended December 31, 2025, the Company recorded a net loss of approximately $
The financial statements do not include any adjustments that might be necessary if the Company is unable to continue as a going concern. Accordingly, the financial statements have been prepared on the basis that assumes the Company will continue as a going concern and which contemplates the realization of assets and satisfaction of liabilities and commitments in the ordinary course of business.
The Company has evaluated the significance of the uncertainty regarding the Company’s financial condition in relation to its ability to meet its obligations, which has raised substantial doubt about the Company’s ability to continue as a going concern. While it is very difficult to estimate the Company’s future liquidity requirements, the Company believes if it is unable to obtain additional financing, existing cash resources will not be sufficient to enable it to fund the anticipated level of operations through one year from the date the accompanying financial statements are issued. There can be no assurances that the Company will be able to secure additional financing on acceptable terms. In the event the Company does not secure additional financing, the Company will be forced to delay, reduce, or eliminate some or all of its discretionary spending, which could adversely affect the Company’s business prospects, ability to meet long-term liquidity needs and the ability to continue operations.
Note 2 – Summary of Significant Accounting Policies
Basis of Presentation and Principles of Consolidation
The accompanying financial statements have been prepared in accordance with the accounting principles generally accepted in the United States of America. The Company’s former wholly-owned subsidiaries, Oxis Biotech, Inc. and Georgetown Translational Pharmaceuticals, Inc., were both dissolved on October 22, 2024. Prior to their dissolution, the operations of Oxis Biotech, Inc. and Georgetown Translational Pharmaceuticals, Inc. were limited with insignificant assets and liabilities.
| 47 |
Accounting Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates include management’s estimates for continued liquidity, accruals for potential liabilities, assumptions used in deriving the fair value of warrant liabilities and Greenshoe Rights liability, valuation of equity instruments issued for debt and services and realization of deferred tax assets.
Cash Equivalents
The
Company considers highly liquid investments with maturities of three months or less at the date of acquisition as cash equivalents
in the accompanying financial statements. As of December 31, 2025 and 2024, total cash equivalents which consist of money market
funds, amounted to approximately $
Restricted Cash
As of December 31, 2025 and 2024, the Company has classified certain cash balances as restricted cash in its balance sheets. The Company’s restricted cash is deposited in a financial institution and held as a collateral for a credit card agreement with the same financial institution.
Deferred Offering Costs
The
Company capitalizes the fair value of equity instruments granted and certain legal, accounting and other third-party fees that are directly
related to the Company’s in-process equity financings until such financings are consummated. After consummation of an equity financing,
these costs are recorded in stockholders’ equity as a reduction of additional paid-in capital generated as a result of the offering.
Should a planned equity financing be abandoned, terminated or significantly delayed, the deferred offering costs are immediately written
off to operating expenses. As of December 31, 2025, there was $
Warrants
The Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s specific terms and applicable authoritative guidance in ASC 480, Distinguishing Liabilities from Equity (“ASC 480”) and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own common stock and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control, among other conditions for equity classification. For issued or modified warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component of additional paid-in capital at the time of issuance. For issued or modified warrants that do not meet all of the criteria for equity classification, the warrants are required to be liability classified and recorded at their initial fair value on the date of issuance and remeasured at fair value at each balance sheet date thereafter. Changes in the estimated fair value of the warrants that are liability classified are recognized as a non-cash gain or loss in the statement of operations at each balance sheet date.
The Company’s use of derivative financial instruments is generally limited to warrants issued by the Company that do not meet the criteria for equity treatment and are recorded as liabilities. We do not use financial instruments or derivatives for any trading purposes.
Greenshoe Rights
In connection with the issuance of the Company’s Series L Convertible Preferred Stock (see Note 5), the Company issued additional investment rights (“Greenshoe Rights”). The Company evaluated the Greenshoe Rights in accordance with ASC 480. As the call option was linked to preferred stock that included redemption features that could require cash settlement upon events outside the Company’s control, the instrument was classified as a liability and initially recognized at fair value. The Greenshoe Rights liability was subsequently remeasured at fair value at each reporting date, with changes in fair value recognized in earnings within other income (expense). Upon execution of the September 2025 waiver eliminating the redemption features, the Greenshoe Rights liability was reclassified to equity at its then-current fair value.
Fair Value of Financial Instruments
Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 820-10 requires entities to disclose the fair value of financial instruments, both assets and liabilities recognized and not recognized on the balance sheet for which it is practicable to estimate fair value. ASC 820-10 defines the fair value of a financial instrument as the amount at which the instrument could be exchanged in a current transaction between willing parties.
| 48 |
The three levels of the fair value hierarchy are as follows:
Level 1 Valuations based on unadjusted quoted prices in active markets for identical assets or liabilities that the entity has the ability to access.
Level 2 Valuations based on quoted prices for similar assets or liabilities, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable data for substantially the full term of the assets or liabilities.
Level 3 Valuations based on inputs that are unobservable, supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The carrying amounts of the Company’s other financial assets and liabilities, such as cash and cash equivalents, short-term investments, prepaid expenses and other current assets, accounts payable, accrued expenses, approximate their
The following table sets forth by level, within the fair value hierarchy, the Company’s liabilities at fair value as of December 31, 2025 and 2024 (as of December 31, 2025 and 2024, no assets were at fair value):
| December 31, 2025 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Liabilities | ||||||||||||||||
| Greenshoe rights liability | $ | $ | $ | $ | ||||||||||||
| Warrant liability | ||||||||||||||||
| Total liabilities | $ | $ | $ | $ | ||||||||||||
| December 31, 2024 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Liabilities | ||||||||||||||||
| Warrant liability | $ | $ | $ | $ | ||||||||||||
| Total liabilities | $ | $ | ||||||||||||||
The change in Level 3 liabilities measured at fair value for the years ended December 31, 2025 and 2024 were as follows:
Warrant Liability
| Warrant Liability | Greenshoe Rights | |||||||
| Balance - December 31, 2023 | $ | $ | ||||||
| Change in fair value of warrant derivative liability | ( | ) | ||||||
| Balance - December 31, 2024 | ||||||||
| Initial recognition of Greenshoe Rights liability | ||||||||
| Change in fair value of warrant derivative liability | ( | ) | ( | ) | ||||
| Reclassification of greenshoe rights liability to equity | ( | ) | ||||||
| Balance - December 31, 2025 | $ | $ | ||||||
| 49 |
Stock-Based Compensation
The Company periodically issues stock-based compensation to officers, directors, employees, and consultants for services rendered. Such issuances vest and expire according to terms established at the issuance date.
Stock-based payments to officers, directors, employees, and consultants in exchange for goods and services, which include grants of employee stock options, are recognized in the financial statements based on their grant date fair values in accordance with ASC 718, Compensation-Stock Compensation. Stock based payments to officers, directors, employees, and consultants, which are generally time vested, are measured at the grant date fair value and depending on the conditions associated with the vesting of the award, compensation cost is recognized on a straight-line or graded basis over the vesting period. Recognition of compensation expense for non-employees is in the same period and manner as if the Company had paid cash for the services. The fair value of stock options granted is estimated using the Black-Scholes option-pricing model, which uses certain assumptions related to risk-free interest rates, expected volatility, expected life, and future dividends. The assumptions used in the Black-Scholes option pricing model could materially affect compensation expense recorded in future periods.
Research and Development Expenses
Costs incurred for research and development are expensed as incurred. The salaries, benefits, and overhead costs of personnel conducting research and development of the Company’s products are included in research and development expenses. Purchased materials that do not have an alternative future use are also expensed. Purchased materials that have an alternative future are classified as a prepaid expense and periodically reviewed.
Basic earnings (loss) per share is computed using the weighted-average number of common shares outstanding during the period. Diluted earnings (loss) per share is computed using the weighted-average number of common shares and the dilutive effect of contingent shares outstanding during the period. Potentially dilutive contingent shares, which primarily consist of stock issuable upon exercise of stock options and warrants, and the conversion of Series L Preferred Stock, have been excluded from the diluted loss per share calculation because their effect is anti-dilutive.
During the year ended December 31, 2025, the Company recorded dividends on Series L Preferred Stock of $
| December 31, 2025 | December 31, 2024 | |||||||
| Series L Preferred Stock 1 | ||||||||
| Options to purchase common stock | ||||||||
| Warrants to purchase common stock | ||||||||
| Total anti-dilutive securities | ||||||||
| 1 |
Concentration
Cash
is deposited in one financial institution. The balances held at this financial institution at times may be in excess of Federal Deposit
Insurance Corporation (“FDIC”) insurance limits of up to $
The Company has a significant concentration of expenses incurred from and accounts payable to Cytovance and the University of Minnesota, see Note 3 – Accounts Payable.
Segment Information
The Company’s Chief Executive Officer and President (“CEO”) is our chief operating decision maker (“CODM”) and evaluates performance and makes operating decisions about allocating resources based on financial data presented on a consolidated basis. Because our CODM evaluates financial performance on a consolidated basis, the Company has determined that it operates as a single reportable segment composed of the financial results of GT Biopharma, Inc. (see Note 8 – Segment Information).
| 50 |
Recent Accounting Pronouncements
In November 2024, the FASB issued ASU 2024-03, Income Statement-Reporting Comprehensive Income-Expense Disaggregation Disclosures (Subtopic 220-40): Disaggregation of Income Statement Expenses, requiring public entities to disclose additional information about specific expense categories in the notes to the financial statements on an interim and annual basis. ASU 2024-03 is effective for fiscal years beginning after December 15, 2026, and for interim periods beginning after December 15, 2027, with early adoption permitted. The Company is currently evaluating the impact of adopting ASU 2024-03.
The Company’s management has evaluated all the recently issued, but not yet effective, accounting standards and guidance that have been issued or proposed by the FASB or other standards-setting bodies through the filing date of these financial statements and does not believe the future adoption of any such pronouncements will have a material effect on the Company’s financial position and results of operations.
Note 3 – Accounts Payable
Accounts payable consists of the following:
| As of | As of | |||||||||||||||
| December 31, 2025 | % | December 31, 2024 | % | |||||||||||||
| Accounts payable to Cytovance | $ | % | $ | % | ||||||||||||
| Accounts payable to University of Minnesota | % | % | ||||||||||||||
| Legal services firm | % | % | ||||||||||||||
| Other accounts payable | % | % | ||||||||||||||
| Total accounts payable | $ | $ | ||||||||||||||
The details of the Company’s accounts payable to Cytovance Biologics, Inc., were as follows:
| Year Ending | ||||||||
| December 31, 2025 | December 31, 2024 | |||||||
| Beginning balance | $ | $ | ||||||
| Invoices, net | ||||||||
| Payments in cash | ( | ) | ( | ) | ||||
| Payments in common stock, at fair value | ( | ) | ||||||
| Payments in pre-funded warrants, at fair value | ( | ) | ||||||
| Ending balance | $ | $ | ||||||
During
the year ended December 31, 2025, the Company issued pre-funded warrants to purchase up to
During the year ended December 31, 2024, the Company issued shares of common stock with a fair value of $ to Cytovance as partial payment of research and development payables. The shares were valued at the respective date of the settlement.
During
the year ended December 31, 2025, a legal services firm currently engaged by the Company agreed to reduce the Company’s prior year
unpaid fees by approximately $
University of Minnesota
See Note 7 – Commitments and Contingencies, Significant Agreements.
PDPC Advisors Inc.
On
June 30, 2025, the Company entered into an Advisory Agreement (the “Advisory Agreement”) with PDPC Advisors Inc.
(“PDPC”), to perform certain advisory services. Under the Advisory Agreement cash payments amounting to $
| 51 |
Note 4 – Warrant Liability
2023 Warrants
On
January 4, 2023, as part of a private placement offering, the Company issued common stock, warrants to purchase up to an aggregate
of
The 2023 Common Warrants and the 2023 Placement Agents Warrants (collectively the “2023 Warrants”), provide for a value calculation for the warrants using the Black Scholes model in the event of certain fundamental transactions. The fair value calculation provides for a floor on the volatility amount utilized in the value calculation at % or greater. The Company has determined this provision introduces leverage to the holders that could result in a value that would be greater than the settlement amount of a fixed-for-fixed option on the Company’s own equity shares. Therefore, pursuant to ASC 815, the Company has classified the 2023 Warrants as a liability in its balance sheet. The classification of the 2023 Warrants, including whether they should be recorded as liability or as equity, is evaluated at the end of each reporting period.
The
2023 Warrants were initially recorded at a fair value at $
The 2023 Warrant liability is valued using a Black Scholes Option pricing model with the following assumptions:
| 2023 Warrants | ||||||||
| December 31, 2025 | December 31, 2024 | |||||||
| Stock price | $ | $ | ||||||
| Risk-free interest rate 1 | % | % | ||||||
| Expected volatility 2 | % | % | ||||||
| Expected life (in years) 3 | ||||||||
| Expected dividend yield 4 | ||||||||
| Fair value of warrants | $ | $ | ||||||
| 1 |
| 2 |
| 3 |
| 4 |
Note 5 – Stockholders’ Equity (Deficit)
The Company’s authorized capital as of December 31, 2025, was shares of common stock, par value $ per share, and shares of preferred stock, par value $ per share.
2025 Private Placement of Equity Facility (the “Committed Equity Facility”)
On
May 14, 2025, and as amended on June 10, 2025, the Company entered into a common shares purchase agreement (the “Common Shares
Purchase Agreement”) with investors (collectively, the “Investors”) relating to the Committed Equity Facility.
Pursuant to the Common Shares Purchase Agreement, the Company has the right from time to time at its option to sell to the Investors
up to $
| 52 |
Sales of the shares of common stock to the Investors under the Common Shares Purchase Agreement, and the timing of any sales, will be determined by the Company from time to time in its sole discretion and will depend on a variety of factors, including, among other things, market conditions, the trading price of the common stock and determinations by the Company regarding the use of proceeds from the sale of such shares of common stock. The net proceeds from any sales under the Common Shares Purchase Agreement will depend on the frequency with, and prices at, which the shares of common stock are sold to the Investors.
The
purchase price of the shares of common stock that the Company elects to sell to the Investors pursuant to the Common Shares Purchase
Agreement will be
In
connection with the execution of the Common Shares Purchase Agreement, the Company agreed to issue to the Investors pre-funded
warrants to purchase an aggregate of
2025 Warrant Inducement Transaction
On
February 26, 2025, the Company received gross proceeds of $
The
Inducement Warrants consist of (i) new Series A Inducement Warrants, representing warrants to purchase up to
The Company determined that under ASC 815, the 2025 inducement and placement agent warrants are considered indexed to the Company’s own stock and eligible for an exception from derivative accounting. Accordingly, the fair value of the 2025 inducement and placement agent warrants are classified as equity.
The
Company recognized the aggregate effect of the modification of warrants and grant of inducement warrants of $
Preferred Stock
Series C Preferred Stock
As of December 31, 2025 and 2024, there were shares of series C preferred stock, par value $ per share (the “Series C Preferred Stock”) issued and outstanding.
As a result of numerous reverse stock-splits in previous years and the agreement terms for adjusting the rights of the related shares, the shares of Series C Preferred Stock are convertible into an infinitesimal amount of common stock, have no voting rights, and in the event of liquidation, the holders of the Series C Preferred Stock would not participate in any distribution of the assets or surplus funds of the Company. The holders of Series C Preferred Stock also are not currently entitled to any dividends if and when declared by the Board. No dividends to holders of the Series C Preferred Stock were declared or unpaid through December 31, 2025.
Series L Preferred Stock and Warrants
As
of December 31, 2025, there were
shares of Series L 10% Convertible Preferred Stock, par value $
per share (the “Series L Preferred Stock”), issued and outstanding that were convertible into
shares of common stock using a conversion price of $
On
May 12, 2025, the Company entered into a securities purchase agreement (as amended on May 21, 2025, the “Securities Purchase
Agreement”) with certain purchasers (the “Purchasers”) providing for the issuance and sale of (i)
shares of the Company’s Series L Preferred Stock with an aggregate stated value of $6,611,111;
(ii) warrants to purchase up to
shares of the Company’s common stock (the “Common Warrants”) with an initial exercise price of $
| 53 |
Pursuant
to the Securities Purchase Agreement, each Purchaser may elect to purchase up to additional shares of Series L
Preferred Stock with an aggregate stated value of up to $
On May 12, 2025, the Company filed a Certificate of Designation of Preferences, Rights and Limitations of Series L 10% Convertible Preferred Stock (the “Certificate of Designation”) with the Secretary of State of the State of Delaware.
Pursuant
to the Securities Purchase Agreement, each Purchaser was issued (i) a Common Warrant and (ii) a Vesting Warrant. An aggregate of
The
shares of Series L Preferred Stock and 2025 Warrants both have full-ratchet price and anti-dilution protections and are subject to
other adjustments, as further described in the Certificate of Designation or the 2025 Warrants, as applicable, subject, solely with
respect to adjustments in connection with the exercise of Greenshoe Rights, to a floor price of $
per share (subject to adjustment for reverse and forward splits, recapitalizations and similar transactions). For the year ended December 31, 2025, the incremental change in the fair value of equity classified instruments
upon reduction of exercise prices was determined to be $
The
Company and the Purchasers entered into a registration rights agreement pursuant to which the Company agreed to file a registration
statement with the Securities and Exchange Commission covering the public resale of the common stock issuable upon conversion of the
Series L Preferred Stock and upon exercise of the 2025 Warrants. The Company agreed to file a registration statement within 30 days
after the initial closing and after each closing of the exercise of a Greenshoe Right in accordance with the Securities Purchase
Agreement, to become effective no later than 90 days after filing. If these deadlines are not met,
Pursuant to the Securities Purchase Agreement, the Company agreed to hold a meeting of its stockholders at the earliest practical date after the execution of the Securities Purchase Agreement for the purpose of obtaining stockholder approval for the issuance, in the aggregate, of more than % of the number of shares of the Company’s common stock outstanding on the date of the initial closing (“Shareholder Approval”). Shareholder Approval was obtained on July 24, 2025.
During
the year ended December 31, 2025, Purchasers elected to exercise their Greenshoe Rights and purchased
additional shares of the Company’s Series L Preferred Stock with a stated value of $
As
of December 31, 2025, dividends to the holders of the Series L Preferred Stock in the amount of $
Series L Convertible Preferred Stock initially classified as Mezzanine Equity
The May 2025 private placement of Series L Preferred Stock and 2025 Warrants described above was allocated to mezzanine equity and additional paid in capital based on the relative fair value of the Series L Preferred Stock and 2025 Warrants at issuance, respectively. Initially, upon the occurrence of certain triggering events, each Purchaser had the right to require the Company to redeem the shares of Series L Preferred Stock, which could require cash settlement upon events outside the Company’s control, as further described in the Certificate of Designation. Because the redemption provisions were not solely within the control of the Company, the Series L Convertible Preferred Stock was initially classified as mezzanine equity in accordance with ASC 480 and ASC 505-10-S99. In September 2025, all the holders of Series L Preferred Stock provided waivers and agreed to waive the rights to redemption set forth in the Certificate of Designation. Accordingly, the Series L Preferred Stock was reclassified from mezzanine equity to shareholders’ equity as it is no longer conditionally redeemable upon the occurrence of certain events that were not solely within the control of the Company.
Greenshoe Rights
The Greenshoe Rights, as additional investment rights
linked to the Series L Convertible Preferred Stock, were initially classified as a liability under ASC 480 due to their association with
the Preferred Stock’s redemption features, which could require cash settlement upon events outside the Company’s control.
They were recognized at fair value of $
| 54 |
Note 6 – Common Stock Warrants and Options
Common Stock Warrants
Stock warrant transactions for the years ended December 31, 2025 and 2024, were as follows:
| Number of | Weighted Average | |||||||
| Warrants | Exercise Price | |||||||
| Warrants outstanding at December 31, 2023 | $ | |||||||
| Granted | ||||||||
| Forfeited/cancelled | ( | ) | ||||||
| Exercised | ||||||||
| Warrants outstanding at December 31, 2024 | $ | |||||||
| Granted | ||||||||
| Anti-dilution adjustment (1) | ||||||||
| Forfeited/cancelled | ( | ) | ||||||
| Exercised | ( | ) | ||||||
| Warrants outstanding at December 31, 2025 | $ | |||||||
| Warrants exercisable at December 31, 2025 | $ | |||||||
| (1) |
The aggregate intrinsic value of all warrants outstanding and all warrants vested and exercisable as of December 31, 2025, was approximately $ million and $ million, respectively, in each case based on the fair value of the Company’s common stock on December 31, 2025.
Pursuant
to the May 2025 private placement of Series L Preferred Stock and 2025 Warrants the Company issued an aggregate of
In
connection with the Committed Equity Facility, the Company issued pre-funded warrants to purchase an aggregate of
In
May 2025 the Company issued warrants underlying
| 55 |
The following table summarizes warrants exercisable as of December 31, 2025:
Range of Exercise Price | Number Outstanding | Weighted Average Remaining Contractual Life (Years) | Weighted Average Exercise Price | |||||||||||
| $ | N/A | $ | ||||||||||||
| N/A | ||||||||||||||
Common Stock Options
In April 2022 the Company established the 2022 Omnibus Incentive Plan (as amended, the “Plan”). The Plan was approved by our Board and stockholders. The purpose of the Plan is to grant stock and options to purchase our common stock, and other incentive awards, to our employees, directors, and key consultants. On July 24, 2025, shareholders voted to increase the maximum number of shares of common stock that may be issued pursuant to awards granted under the Plan by . Pursuant to the increase, the maximum number of shares of common stock that may be issued pursuant to awards granted under the Plan is . The shares of our common stock underlying cancelled and forfeited awards issued under the Plan may again become available for grant under the Plan. As of December 31, 2025, there were stock options outstanding and shares of restricted stock granted in prior years under the Plan, which left shares available for grant under the Plan.
| Number of | Weighted Average | |||||||
| Options | Exercise Price | |||||||
| Options outstanding at December 31, 2023 | $ | |||||||
| Granted | ||||||||
| Forfeited/cancelled | ( | ) | ||||||
| Exercised | ||||||||
| Options outstanding at December 31, 2024 | $ | |||||||
| Granted | ||||||||
| Forfeited/cancelled | ( | ) | ||||||
| Exercised | ||||||||
| Options outstanding at December 31, 2025 | $ | |||||||
| Options exercisable at December 31, 2025 | $ | |||||||
The weighted average remaining contractual life of all options outstanding, and all options vested and exercisable as of December 31, 2025, was approximately years. Furthermore, the aggregate intrinsic value of all options outstanding and all options vested and exercisable as of December 31, 2025, was $, in each case based on the fair value of the Company’s common stock on December 31, 2025.
The total fair value of options that vested during years ended December 31, 2025 and 2024, was $ and $, respectively, and is included in selling, general and administrative expense in the accompanying statements of operations. As of December 31, 2025, stock options were vested and exercisable and unvested compensation expense amounted to approximately $.
On August 19, 2025, the Company granted stock options to directors and officers to purchase an aggregate of shares of common stock. The stock options are exercisable at $ per share, expire in years, vest either immediately or over the remainder of 2025 and 2026, with a fair value of approximately $ on the date of grant which will be amortized over the vesting period.
| 56 |
On October 17, 2024, the Company granted stock options to an officer to purchase an aggregate of shares of common stock. The stock options are exercisable at $ per share, expire in years, vest over three years with a fair value of approximately $ on the date of grant which will be amortized over the vesting period.
Range of Exercise Price | Number Outstanding | Weighted Average Remaining Contractual Life (Years) | Weighted Average Exercise Price | |||||||||||
| $ | $ | |||||||||||||
Note 7 – Commitments and Contingencies
Litigation
The Company is involved in certain legal proceedings that arise from time to time in the ordinary course of our business. Except for income tax contingencies, we record accruals for contingencies to the extent that our management concludes that the occurrence is probable and that the related amounts of loss can be reasonably estimated. Legal expenses associated with the contingency are expensed as incurred. There is no current or pending litigation of any significance with the exception of the matters identified below that have arisen under, and are being handled in, the normal course of business:
Ohri Matter
On July 22, 2024, the Company filed an AAA Arbitration Demand against Manu Ohri, its former Chief Financial Officer. In the demand, the Company asserts claims against Mr. Ohri for breach of his fiduciary duties and breach of contract and seeks a declaratory judgment providing that the Company may characterize Mr. Ohri’s termination as “for cause” under his employment agreement, and that the Company may revoke the separation agreement entered into between the Company and Mr. Ohri prior to the Company learning of Mr. Ohri’s breaches. In addition to the declaratory judgment, the Company seeks damages arising from Mr. Ohri’s violations, and attorneys’ fees and any forum and arbitration fees. On September 3, 2024, Mr. Ohri filed both a general denial of the Company’s claims against him and counterclaims for breach of his employment agreement and separation agreement. The final hearing date, originally scheduled for June 10, 2025, has been postponed indefinitely in order to allow the parties to mediate the dispute. Mediation is scheduled for March 31, 2026. Should mediation be unsuccessful, the case will proceed to a final hearing in October 2026. At this stage in the proceedings the Company is not able to determine the probability of the outcome of this matter or a range of reasonably expected losses, if any. The Company believes it has recorded an appropriate accrual for the matter.
TWF Global Matter
On
May 24, 2023, TWF Global, LLC (“TWF”) filed a Complaint in the California Superior Court for the County of Los Angeles naming
the Company as defendant. The complaint alleges that TWF is the holder of two Convertible Promissory Notes (“Notes”) and
that the Company did not deliver shares of common stock due on conversion in February 2021. TWF was seeking per diem liquidated damages
based on the terms of alleged Notes. On July 14, 2023, the Company filed a motion to dismiss for improper forum because the terms of
the Notes, as alleged, require disputes to be filed in New York state and federal courts. TWF voluntarily dismissed its complaint before
the California Superior Court of Los Angeles without prejudice. The Company subsequently filed a summons and complaint for interpleader
against TWF and Z-One, LLC before the Supreme Court of the State of New York County of New York, asking the Supreme Court to determine
if the Company’s shares of common stock should be registered to TWF or Z-One LLC, as both of these entities have made conflicting
demands for the shares. On February 5, 2024, the Company filed a motion for entry of default against TWF, seeking an order directing
the Company to register the shares of common stock in the name of Z-One, LLC and that the Company be released from all associated liability
and claims. The Court denied the motion without prejudice and agreed to reconsider the motion without further briefing upon the filing
of a supplemental party affidavit. On May 9, 2024, Z-One, LLC filed a motion for summary judgement seeking dismissal of the action, representing
that Z-One, LLC and TWF have settled their dispute over the entitlement to the Company’s shares of common stock and there is no
remaining dispute before the Court. On May 21, 2024, the Company filed a supplemental affidavit in support of its motion for entry of
default. On November 14, 2024, the Court held a hearing on the parties’ motions, at which the Court found that the motion for entry
of default was mooted by the settlement agreement between Z-One, LLC and TWF. The Court ordered that the case be dismissed. On February
17, 2025, Z-One, LLC filed a Summons with Notice in the Supreme Court of the State of New York, County of New York. The Company then
filed a demand that Z-One, LLC serve a complaint, and on June 25, 2025, Z-One, LLC filed a Complaint alleging that it is the holder,
either originally or by assignment, of a Convertible Note in the principal amount of $
| 57 |
Silberfein, DiPietro, and Werthman Trust Matters
On
July 8, 2025, Coby Silberfein filed a summons with notice in the State of New York Supreme Court for the County of New York naming the
Company as defendant seeking damages for breach of a securities purchase agreement and convertible note in the principal amount of $
Significant Agreements
Cytovance Biologics, Inc.
In October 2020, the Company entered into a Master Services Agreement with Cytovance Biologics, Inc. (“Cytovance”), to perform biologic development and manufacturing services, and to produce and test compounds used in the Company’s potential product candidates. The Company subsequently executed numerous Statements of Work (“SOWs”) for the research and development of products for use in clinical trials.
On
August 24, 2022, the Company entered into a Settlement and Investment Agreement with Cytovance that amended existing SOWs and allowed
for future invoices to be settled in in a combination of cash and issuance of the Company’s common stock. The Agreement also set
Cytovance’s beneficial ownership limitation at
On
April 25, 2024, the Company entered into an Amendment to the Settlement and Investment Agreement with Cytovance that increased Cytovance’s
beneficial ownership limitation to
During
the years ended December 31, 2025 and 2024, the Company recognized research and development expenses of $
As
of December 31, 2025, the Company’s commitments in relation to unbilled and unaccrued SOWs and any related Change Orders from Cytovance
for services that have not yet been rendered as of December 31, 2025, amounted to approximately $
University of Minnesota
2023 Sponsored Research Agreement
On
May 20, 2024, the Company entered into a sponsored research agreement (the “2023 Sponsored Research Agreement”) with the
Regents of the University of Minnesota (the “University of Minnesota”), effective July 1, 2023. Payments totaling approximately $
| 58 |
On
June 18, 2025, the 2023 Sponsored Research Agreement was amended to expire on December 31, 2025. In addition, payments amounting to $
The
Company recorded an expense classified as research and development of approximately $
As of December 31, 2025, there were no outstanding commitments in relation to unbilled and unaccrued amounts from the University of Minnesota pursuant to the 2023 Sponsored Research Agreement for services that have not yet been rendered as of December 31, 2025.
2016 Exclusive Patent License Agreement
Effective
July 18, 2016, the Company entered into an exclusive patent license agreement with the University of Minnesota (as amended,
the “2016 Exclusive Patent License Agreement”), to further develop and commercialize cancer therapies using TriKE®
technology developed by researchers at the University of Minnesota to target NK cells to cancer. Under the terms of the agreement, the
Company receives exclusive rights to conduct research and to develop, make, use, sell, and import TriKE® technology worldwide
for the treatment of any disease, state, or condition in humans. The Company is responsible for obtaining all permits, licenses, authorizations,
registrations, and regulatory approvals required or granted by any governmental authority anywhere in the world that is responsible for
the regulation of products such as the TriKE® technology, including without limitation the FDA and the European Agency
for the Evaluation of Medicinal Products in the European Union. The agreement requires an upfront payment of $
Effective
May 13, 2024, the Company entered into an amended and restated exclusive patent license agreement with the University of Minnesota (the “A&R 2016 Exclusive Patent License Agreement”).
The amendment requires an upfront payment of $
The
Company recorded an expense classified as research and development of $
2021 Exclusive License Agreement
Effective
March 26, 2021, the Company entered into an exclusive license agreement with the University of Minnesota (the “2021 Exclusive
Patent License Agreement”), specific to the B7H3 targeted TriKE®. The agreement requires an upfront payment of
$
| 59 |
The
Company recorded an expense classified as research and development of $
2024 GTB-3650 Clinical Trial Agreement
On
November 18, 2024, the Company entered into an investigator initiated clinical trial agreement (the “2024 Clinical Trial
Agreement”) with the University of Minnesota, pursuant to which, the University of Minnesota shall sponsor an IND application
for IND 165546 GTB-3650 (the “Research Program”) and shall serve as a sponsor investigator for a phase 1 clinical trial
entitled, “GTB-3650 (CD16/IL-15/CD33) Tri-Specific Killer Engager (TriKE) for the Treatment of High Risk Myelodysplastic
Syndromes (MDS), Refractory/Relapsed Acute Myeloid Leukemia (AML), and Minimal Residual Disease in AML,” designed by the
University of Minnesota (the “Study”). The Research Program is being conducted for clinical research use. The budget for
the Study, including without limitations, funding and resources, provides for up to approximately $
The
Company recorded an expense classified as research and development of approximately $
As
of December 31, 2025, the Company’s commitments in relation to unbilled and unaccrued amounts from the University of Minnesota
pursuant to the 2024 Clinical Trial Agreement for services that have not yet been rendered as of December 31, 2025, amounted to approximately
$
Contingency – NASDAQ Matters
On
November 21, 2024, we received a letter (the “Notification Letter”) from the Nasdaq Listing Qualifications Staff (the “Staff”)
notifying us that the amount of our stockholders’ equity had fallen below the $
The Notification Letter indicated that we had 45 days (i.e., until January 6, 2025) to submit a plan to regain compliance with the Minimum Stockholders’ Equity Requirement, noting that if such plan is accepted, the Staff can grant us an extension of up to 180 days from the date of the Notification Letter to evidence compliance. In determining whether to accept our plan, the Staff will consider such things as the likelihood that the plan will result in compliance with Nasdaq’s continued listing criteria, our past compliance history, the reasons for our current non-compliance, other corporate events that may occur within the review period, our overall financial condition and our public disclosures.
We submitted a plan of compliance to the Staff on December 31, 2024, outlining our plan to conduct periodic public and private securities offerings to regain compliance. The Staff also requested additional information regarding our financing plans and financial projection, which we provided to them. On June 13, 2025, we received a letter from the Staff notifying us that we had regained compliance with the Minimum Stockholders’ Equity Requirement set forth in Nasdaq Listing Rule 5550(b)(1). Nasdaq will continue to monitor the Company to ensure its ongoing compliance with the Minimum Stockholders’ Equity Requirement, so if at the time of the filing of the Company’s next periodic financial statements the Company does not evidence compliance with the Minimum Stockholders’ Equity Requirement, the Company may be subject to delisting.
On
November 20, 2025, the Company received a letter (the “2025 Letter”) from the Staff of The Nasdaq Stock Market LLC (“Nasdaq”) notifying the Company that its common stock, $
par value per share
In accordance with Nasdaq Listing Rule 5810(c)(3)(A), the Company was provided a compliance period of 180 calendar days from the date of the Letter, or until May 19, 2026 (the “Compliance Period”), to regain compliance with the Minimum Bid Price requirement. If at any time during the Compliance Period, the closing bid price of the Company’s common stock is at least $ per share for a minimum of ten consecutive business days (unless the Nasdaq staff exercises its discretion to extend this ten business day period pursuant to Nasdaq Listing Rule 5810(c)(3)(H)), Nasdaq will provide the Company written confirmation of compliance with the Minimum Bid Price, and the matter will be closed.
| 60 |
If the Company does not regain compliance during the Compliance Period, the Company may be eligible for an additional 180-calendar day period to regain compliance with the Minimum Bid Price, provided that it meets the applicable market value of publicly held shares requirement for continued listing and all other applicable standards for initial listing on The Nasdaq Capital Market (except the Minimum Bid Price requirement), and notifies Nasdaq of its intent to cure the deficiency by effecting a reverse stock split of its common stock, if necessary. If Nasdaq determines that the Company is not eligible for an additional 180 calendar days compliance period or the Company will not be able to cure the deficiency with the Minimum Bid Price requirement within the allotted compliance period, the Company’s stock will be subject to delisting.
The Company intends to monitor the closing bid price of the common stock and assess its available options to regain compliance with the Minimum Bid Price requirement and continue listing on The Nasdaq Capital Market. There can be no assurance that the Company will be able to regain compliance with the Minimum Bid Price requirement or will otherwise be in compliance with other applicable Nasdaq listing rules.
If our common stock is delisted from Nasdaq, our ability to raise capital through public offerings of our securities and to finance our operations could be adversely affected. We also believe that delisting would likely result in decreased liquidity and/or increased volatility in our common stock and could harm our business and future prospects. In addition, we believe that, if our common stock is delisted, our stockholders would likely find it more difficult to obtain accurate quotations as to the price of our common stock and it may be more difficult for stockholders to buy or sell our common stock at competitive market prices, or at all.
Note 8 – Segment Information
The
Company operates and manages its business as
The Company’s CODM reviews financial information presented and decides how to allocate resources based on net income (loss). Net income (loss) is used for evaluating financial performance.
Significant segment expenses include research and development, salaries, insurance, and stock-based compensation. Operating expenses include all remaining costs necessary to operate our business, which primarily include external professional services and other administrative expenses. The following table presents the significant segment expenses and other segment items regularly reviewed by our CODM:
| Year Ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Research and development | $ | $ | ||||||
| Salaries | ||||||||
| Insurance | ||||||||
| Stock-based compensation | ||||||||
| Operating expenses | ||||||||
| Other (income) expense | ||||||||
| Net loss | $ | $ | ||||||
| 61 |
Note 9 – Income Tax
The
Company did not record any income tax provision for the years ended December 31, 2025 and 2024, respectively, due to the Company’s
net losses. The Company files income tax returns in the United States (“Federal”) and California and Minnesota (“State”)
jurisdictions. The Company is subject to Federal and State income tax examinations by tax authorities for all years since its inception.
At December 31, 2025, the Company had Federal and State net operating loss carry forwards available to offset future taxable income of
approximately $
Based
on the weight of available evidence, including cumulative losses in recent years and expectations of future taxable income, the Company
has determined that it was more likely than not that its deferred tax assets would not be realized at December 31, 2025 and 2024, respectively.
Accordingly, the Company has recorded a valuation allowance for
Deferred taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes, and operating losses and tax credit carryforwards. The significant components of net deferred income tax assets are:
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| Deferred tax assets: | ||||||||
| Federal net operating loss carryforward | $ | $ | ||||||
| Stock based compensation and other items | ||||||||
| Intellectual property | ||||||||
| Section 14 research and development | ||||||||
| Deferred tax assets before valuation | ||||||||
| Valuation allowance | ( | ) | ( | ) | ||||
| Net deferred income tax assets | $ | $ | ||||||
A reconciliation of the federal statutory income tax rate and the effective income tax rate as a percentage of income before income tax provision is as follows for the years ended:
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| Federal statutory income tax rate | % | % | ||||||
| State tax, net of federal benefit | % | % | ||||||
| Change in valuation allowance on net operating loss carryforwards | ( | )% | ( | )% | ||||
| Effective income tax rate | % | % | ||||||
Note 10 – Restatement of Quarterly Financial Statements (Unaudited)
As further described below, our unaudited financial statements covering the quarterly reporting periods during fiscal year 2025, consisting of the quarters ended June 30, 2025, and September 30, 2025 have been restated to reflect the correction of material errors.
Restatement Background
The need for the restatement arose out of the results of certain financial analysis the Company performed in the course of preparing for the audit of its December 31, 2025 financial statements. Upon reevaluation of the Greenshoe Rights connected to the Series L Preferred Stock issued in the May 2025 private placement, it was determined that the Greenshoe Rights are purchase options of the Series L 10% Convertible Preferred Stock, the Greenshoe Rights met the criteria for liability classification in accordance with ASC 480 because they are exercised into Series L 10% Convertible Preferred Stock that is redeemable upon triggering events outside the issuer’s control. Accordingly, the Greenshoe Rights required classification as a liability and marked to market at each reporting date as required under GAAP. Therefore, the Company recognized other income (expense) for the change in the fair value of the Greenshoe Rights liability at each reporting date, and reclassified the Greenshoe Rights liability to equity as of the date of the redemption rights waiver.
Restatement Adjustments
The Company inadvertently did not classify the Greenshoe Rights as a liability, which led to accounting adjustments to correct the errors identified. The Company is providing restated quarterly and year-to-date unaudited financial information for June 30, 2025, and September 30, 2025, in its audited financial statements for the year ended December 31, 2025. The following table summarizes the effect of the errors on the Company’s balance sheets as of June 30, 2025, and September 30, 2025, and on the respective statements of operations and statements of cash flows.
| 62 |
The restated balance sheet line items for the second and third fiscal quarters of 2025 are as follows:
| June 30, 2025 (Unaudited) | ||||||||||||
| Originally Reported | Adjustment | Restated | ||||||||||
| Total Assets | $ | $ | $ | |||||||||
| Greenshoe Rights liability | ||||||||||||
| Total Liabilities | ||||||||||||
| Mezzanine Equity | ||||||||||||
| Convertible Preferred stock | ||||||||||||
| Common stock | ||||||||||||
| Additional paid in capital | ||||||||||||
| Accumulated deficit | ( | ) | ( | ) | ( | ) | ||||||
| Total Stockholders’ Equity (Deficit) | ( | ) | ( | ) | ||||||||
| Total Liabilities and Stockholders’ Equity (Deficit) | $ | $ | $ | |||||||||
| September 30, 2025 (Unaudited) | ||||||||||||
| Originally Reported | Adjustment | Restated | ||||||||||
| Total Assets | $ | $ | $ | |||||||||
| Greenshoe Rights liability | ||||||||||||
| Total Liabilities | ||||||||||||
| Convertible Preferred stock | ||||||||||||
| Common stock | ||||||||||||
| Additional paid in capital | ||||||||||||
| Accumulated deficit | ( | ) | ( | ) | ( | ) | ||||||
| Total Stockholders’ Equity (Deficit) | ||||||||||||
| Total Liabilities and Stockholders’ Equity (Deficit) | $ | $ | $ | |||||||||
The restated line items of the statements of operations for the three months ended June 30, 2025, and September 30, 2025, are as follows:
| Three Months Ended June 30, 2025 (Unaudited) | ||||||||||||
| Originally Reported | Adjustment | Restated | ||||||||||
| Loss from Operations | $ | ( | ) | $ | $ | ( | ) | |||||
| Loss on initial recognition of Greenshoe Rights liability | ( | ) | ( | ) | ||||||||
| Total Other Income (Expense) | ( | ) | ( | ) | ||||||||
| Net Loss | ( | ) | ( | ) | ( | ) | ||||||
| Dividend on Series L Preferred Stock | ( | ) | ( | ) | ||||||||
| Net Loss attributable to common stockholders’ | ( | ) | ( | ) | ( | ) | ||||||
| Net Loss Per Share - Basic and Diluted | $ | ) | $ | ) | $ | ) | ||||||
| Weighted average common shares outstanding - basic and diluted | ||||||||||||
| Three Months Ended September 30, 2025 (Unaudited) | ||||||||||||
| Originally Reported | Adjustment | Restated | ||||||||||
| Loss from Operations | $ | ( | ) | $ | $ | ( | ) | |||||
| Change in fair value of Greenshoe Rights liability | ||||||||||||
| Total Other Income (Expense) | ||||||||||||
| Net Loss | ( | ) | ||||||||||
| Dividend on Series L Preferred Stock | ( | ) | ( | ) | ||||||||
| Deemed dividend | ( | ) | ( | ) | ||||||||
| Net Loss attributable to common stockholders’ | ( | ) | ||||||||||
| Net Loss Per Share - Basic and Diluted | $ | ) | $ | $ | ||||||||
| Weighted average common shares outstanding - basic and diluted | ||||||||||||
The restated line items of the statements of operations for the six months ended June 30, 2025; and the nine months ended September 30, 2025, are as follows:
| Six Months Ended June 30, 2025 (Unaudited) | ||||||||||||
| Originally Reported | Adjustment | Restated | ||||||||||
| Loss from Operations | $ | ( | ) | $ | $ | ( | ) | |||||
| Loss on initial recognition of Greenshoe Rights liability | ( | ) | ( | ) | ||||||||
| Total Other Income (Expense) | ( | ) | ( | ) | ||||||||
| Net Loss | ( | ) | ( | ) | ( | ) | ||||||
| Dividend on Series L Preferred Stock | ( | ) | ( | ) | ||||||||
| Net Loss attributable to common stockholders’ | ( | ) | ( | ) | ( | ) | ||||||
| Net Loss Per Share - Basic and Diluted | $ | ) | $ | ) | $ | ) | ||||||
| Weighted average common shares outstanding - basic and diluted | ||||||||||||
| Nine Months Ended September 30, 2025 (Unaudited) | ||||||||||||
| Originally Reported | Adjustment | Restated | ||||||||||
| Loss from Operations | $ | ( | ) | $ | $ | ( | ) | |||||
| Loss on initial recognition of Greenshoe Rights liability | ( | ) | ( | ) | ||||||||
| Change in fair value of Greenshoe Rights liability | ||||||||||||
| Total Other Income (Expense) | ( | ) | ( | ) | ||||||||
| Net Loss | ( | ) | ( | ) | ( | ) | ||||||
| Dividend on Series L Preferred Stock | ( | ) | ( | ) | ||||||||
| Deemed dividend | ( | ) | ( | ) | ||||||||
| Net Loss attributable to common stockholders’ | ( | ) | ( | ) | ( | ) | ||||||
| Net Loss Per Share - Basic and Diluted | $ | ) | $ | ) | $ | ) | ||||||
| Weighted average common shares outstanding - basic and diluted | ||||||||||||
| 63 |
While the adjustments changed net loss and added a change in fair value of Greenshoe Rights in the statements of cash flow statements, they did not have an impact on total net cash provided by operating activities, net cash used in investing activities, or net cash provided by (used in) financing activities for any of the applicable periods.
The restated line items of the statements of cash flows for the six months ended June 30, 2025, and the nine months ended September 30, 2025, are as follows:
| Six Months Ended June 30, 2025 (Unaudited) | ||||||||||||
| Originally Reported | Adjustment | Restated | ||||||||||
| Net Loss | $ | ( | ) | $ | ( | ) | $ | ( | ) | |||
| Loss on initial recognition of Greenshoe Rights liability | ||||||||||||
| Net cash used in operating activities | $ | ( | ) | $ | $ | ( | ) | |||||
| Nine Months Ended September 30, 2025 (Unaudited) | ||||||||||||
| Originally Reported | Adjustment | Restated | ||||||||||
| Net Loss | $ | ( | ) | $ | ( | ) | $ | ( | ) | |||
| Loss on initial recognition of Greenshoe Rights liability | ||||||||||||
| Change in fair value of Greenshoe Rights liability | ( | ) | ( | ) | ||||||||
| Net cash used in operating activities | $ | ( | ) | $ | $ | ( | ) | |||||
| SUPPLEMENTAL DISCLOSURES OF NON-CASH INVESTING AND FINANCING ACTIVITIES | ||||||||||||
| Reclassification of Greenshoe Rights liability to equity | $ | $ | ||||||||||
Note 11 – Subsequent Events
Exercise of Series L 10% Convertible Preferred Stock Greenshoe Rights
From
January 1, 2026 through March 23, 2026, Purchasers of Series L Preferred Stock elected to exercise their Greenshoe Rights and purchased shares of the Series L Preferred Stock with a stated
value of $
Conversion of Series L Preferred Stock
From January 1, 2026 through March 23, 2026, the holders of the Company’s Series L Preferred Stock have converted shares of Series L Preferred Stock into shares of common stock.
Exercise of Warrants
From January 1, 2026 through March 23, 2026, warrants
underlying shares of Company’s common stock were exercised resulting in net proceeds of approximately
| 64 |
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
There were no changes in or disagreements with our accountants on accounting and financial disclosure during the last two fiscal years.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. For purposes of this section, the term “disclosure controls and procedures” means controls and other procedures of an issuer that are designed to ensure that information required to be disclosed by the issuer in the reports that it files or submits under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by an issuer in the reports that it files or submits under the Exchange Act is accumulated and communicated to the issuer’s management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of December 31, 2025, the end of the period covered by this report, our disclosure controls and procedures were not effective at a reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Securities Exchange Act of 1934, as amended, as a process designed by, or under the supervision of, a company’s principal executive and principal accounting officers and effected by a company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| ● | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the Company | |
| ● | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of the financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and | |
| ● | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements. |
All internal control systems, no matter how well designed, have inherent limitations and can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within our company have been detected. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Management evaluated the effectiveness of our internal control over financial reporting as of December 31, 2025, using the framework set forth in the report of the Treadway Commission’s Committee of Sponsoring Organizations (“COSO”),“2013 Internal Control– Integrated Framework.” Based upon that evaluation, management believes our internal control over financial reporting was not effective as of December 31, 2025.
Subsequent to filing our Form 10-Q for the third quarter ended September 30, 2025, and as a result of additional analysis performed in preparation for the December 31, 2025 audit, management determined that the Company did not maintain effective controls over the preparation and review of accounting for complex financial transactions, specifically with respect to the proper application, of ASC 480 Distinguishing Liabilities from Equity (“ASC 480”) related to certain stock purchase rights (“Greenshoe Rights”). As the Greenshoe Rights are purchase options of the Series L 10% Convertible Preferred Stock, the Greenshoe Rights should be classified as liability in accordance with ASC 480 because they are exercised into Series L 10% Convertible Preferred Stock that is redeemable upon triggering events outside the issuer’s control. Accordingly, the Greenshoe Rights required classification as a liability and marked to market at each reporting date as required under GAAP in the quarterly periods ended June 30, 2025, and September 30, 2025. This resulted in an error in our interim quarterly financial statements as originally reported in our Quarterly Reports on Form 10-Q for the quarterly periods ended June 30, 2025, and September 30, 2025, which in turn required a restatement of our interim financial data for those periods within this Annual Report on Form 10-K. Management determined that this control deficiency constituted a material weakness in internal control over financial reporting as of June 30, 2025, and September 30, 2025. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis.
Inherent Limitations on the Effectiveness of Controls
Management does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent or detect all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control systems are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in a cost-effective control system, no evaluation of internal control over financial reporting can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, have been or will be detected.
These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of a simple error or mistake. Controls can also be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls is based in part on certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Projections of any evaluation of controls effectiveness to future periods are subject to risks. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures.
Changes in Internal Controls Over Financial Reporting
There were no other changes in our internal control over financial reporting identified in connection with the evaluation required by paragraph (d) of Rule 13a-15 or Rule 15d-15 under the Exchange Act that occurred during or subsequent to the Company’s last fiscal quarter of the period covered by this report that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Remediation of Material Weakness
Subsequent to September 30, 2025, the Company adopted additional internal controls wherein if the issuance of stock purchase rights or other derivative financial instruments, or any other complex transaction is contemplated, an accounting consultant will be engaged as to the financial statement impact that any such transaction may have, prior to consummation of the transaction.
ITEM 9B. OTHER INFORMATION
Rule 10b5-1 Trading Plans
During
the quarter ended December 31, 2025, no director or officer of the Company
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
| 65 |
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The following table sets forth the name, age, position and date of appointment of each of our directors and executive officers as of February 23, 2026.
| Name | Age | Position | Date of Appointment | |||
| Michael Breen | 63 | Executive Chairman of the Board and Chief Executive Officer | January 13, 2021 | |||
| Alan Urban | 57 | Chief Financial Officer & Secretary | June 3, 2024 | |||
| Charles J. Casamento (1) (2) | 80 | Director | May 1, 2023 | |||
| Hilary Kramer (2) (3) | 60 | Director | May 7, 2025 | |||
| David C. Mun-Gavin (2) (4) | 79 | Director | June 10, 2025 |
| (1) | Chairman of the Audit Committee. | |
| (2) | Member of the Audit Committee, the Compensation Committee and the Nominating and Corporate Governance Committee. | |
| (3) | Chairman of the Nominating and Corporate Governance Committee. | |
| (4) | Chairman of the Compensation Committee. |
Michael Breen – Executive Chairman of the Board and Chief Executive Officer
Mr. Breen was appointed to our Board of Directors on January 13, 2021, was appointed Executive Chairman of the Board on November 8, 2021, was appointed as our Interim Chief Executive Officer on March 2, 2022, and was appointed as our Chief Executive Officer on April 29, 2025. Prior to joining our company, Mr. Breen served as a senior partner in the global law firm of Clyde & Co., specializing in all aspects of corporate law, including mergers and acquisitions and fund management regulatory issues, which included advising clients in the biotechnology and health sciences sectors. Prior to joining Clyde & Co., Mr. Breen served as a senior partner and managing partner in the London law firm of Edward Lewis. Prior to his time at Edward Lewis, he was also a partner at Robert Gore & Company. Between 2002 and 2005, Mr. Breen was managing director and a shareholder of the Sports and Entertainment Division of Insinger de Beaufort Bank, a Dutch private banking, asset management and trust group listed on the Luxembourg stock exchange. From 2001 to 2007 Mr. Breen also served as a non-executive director and co-owner of Damon Hill Holdings Limited, a multi franchise motor dealer group. Mr. Breen also serves as a director of a Cayman Islands fund, Bristol Investment Fund, Limited. Mr. Breen is also a non-executive director and co-owner of Colorsport Images Limited, a sports photographic agency and library. Mr. Breen is a U.K. qualified solicitor/attorney who holds an Honors LL.B. degree in law from the University College of Wales, Aberystwyth and qualified as a solicitor of the Supreme Court of Judicature of England and Wales in 1988. Mr. Breen is a former member of the International Bar Association, British Association for Sport and the Law, Law Society of England and Wales, and Holborn Law Society. We believe that Mr. Breen is well-qualified to serve as a director due to his demonstrated leadership abilities and extensive experience serving on the boards of directors of numerous corporations.
Alan Urban – Chief Financial Officer
Mr. Urban has previously served as a member of the Board of Directors of the Company from June 2022 to May 2023; as Chief Financial Officer for SRAX, Inc. (OTC: SRAX), a financial technology company, from March 2023 to July 2023; as Chief Financial Officer for Creek Road Miners, Inc. (formerly OTC: CRKR), a cryptocurrency mining company, from November 2021 to March 2023; and as Chief Financial Officer and Secretary for Research Solutions, Inc. (NASDAQ: RSSS), a SaaS and content provider in the scientific, technical and medical information space, from October 2011 to October 2021. Earlier in his career, Mr. Urban served as Chief Financial Officer and Senior Vice President of Finance and Accounting for ReachLocal, Inc. (NASDAQ: RLOC), an internet marketing company; and as Vice President of Finance and Treasurer for Infotrieve, Inc., a content provider in the scientific, technical and medical information space. He has been a Certified Public Accountant (currently inactive) since 1998. Mr. Urban received a B.S. in Business, with a concentration in Accounting Theory and Practice, from California State University, Northridge.
Charles J. Casamento - Director
Mr. Casamento was appointed to our Board of Directors on May 1, 2023. Mr. Casamento is currently executive director and principal of The Sage Group, a healthcare advisory group specializing in mergers, acquisitions, and partnerships between biotechnology companies and pharmaceutical companies, since 2007. He was the president and CEO of Osteologix, Inc., a public biopharmaceutical company developing products for treating osteoporosis, from 2004 through 2007. Mr. Casamento was founder of, and from 1999 through 2004, served as chairman of the board, president and CEO, of Questcor Pharmaceuticals, Inc. which was subsequently acquired by Mallinckrodt Pharmaceuticals. Mr. Casamento formerly served as RiboGene, Inc.’s president, CEO and chairman of the board from 1993 through 1999 until it merged with Cypros Pharmaceutical Corp to form Questcor Pharmaceuticals, Inc. He was co-founder, president and CEO of Interneuron Pharmaceuticals, Inc. (Indevus), a biopharmaceutical company, from 1989 until 1993. Indevus was eventually acquired by Endo Pharmaceuticals. Mr. Casamento has also held senior management positions at Genzyme Corporation, where he was senior vice president, pharmaceuticals and biochemicals; American Hospital Supply, where he was vice president of business development and strategic planning for the Critical Care Division; Johnson & Johnson, Hoffmann-LaRoche, Inc. and Sandoz Inc. (now Novartis). Mr. Casamento also serves on the board of directors of the following Nasdaq listed companies: Eton Pharmaceuticals, Inc., PaxMedica, Inc. and Relmada Therapeutics, Inc. During his career he has served on the boards of fourteen Biotech/Pharma companies and has also been a director and vice chairman of The Catholic Medical Missions Board, a large not-for-profit organization providing health care services to third world countries. He has served as a guest lecturer at Fordham University and is on the Science Council of Fordham University. He holds a bachelor’s degree in Pharmacy from Fordham University and an MBA from Iona University and was originally licensed to practice pharmacy in the states of New York and New Jersey. We believe that Mr. Casamento is well-qualified to serve as a director due to his extensive experience with biotechnology and pharmaceutical companies, and serving on the boards of directors of numerous publicly traded corporations.
| 66 |
Hilary Kramer - Director
Ms. Kramer was appointed to our Board of Directors on May 7, 2025. Ms. Kramer has served as the Chief Executive Officer, President and Chief Investment Officer of Greentech Research LLC, a global fund specializing in alternative energies and clean technologies, since 2006. Prior to this, she served as an analyst and investment banker at each of Morgan Stanley and Lehman Brothers. Ms. Kramer previously served as the co-head and a director of Ibero-American Media Partners II Ltd., a $1.0 billion private equity fund jointly owned by Hicks, Muse, Tate & Furst. Ms. Kramer has served as a director of multiple publicly-traded and privately owned companies, including INX Digital Company, RSL Communications, Inc., El Sitio Inc., Deltathree Inc. and Direct TV Latin America. She has also consulted with family offices and institutions, such as Montgomery Asset Management, Freddie Mac, and families on the Forbes list of global billionaires ranging from Latin America to the Middle East. Ms. Kramer authors the futurist and investment newsletter, GameChangers, and has a nationally syndicated investment radio show on the Salem Network in 140 markets. She is the author of Ahead of the Curve (Simon & Schuster 2007) and The Little Book of Big Profits from Small Stocks (Wiley 2012) and Game Changing Investing (Regnery 2020). Ms. Kramer was a founding member of the Wall Street Journal Women in Business. Ms. Kramer holds an MBA from the Wharton School of the University of Pennsylvania and a BA with honors from Wellesley College. We believe that Ms. Kramer is well-qualified to serve as a director due to her extensive experience serving on the boards of directors of numerous corporations.
David C. Mun-Gavin - Director
Mr. Mun-Gavin was appointed to our Board of Directors on June 10, 2025. Mr. Mun-Gavin has served as the Client Director, International Private Banking Division, of Brown Shipley, a Quintet private bank, since October, 2018. Prior to this, Mr. Mun-Gavin served in various roles at Insinger De Beaufort, Credit Suisse, The Vanol Group, Swiss Bank Corporation Investment Banking Ltd., Bankers Trust International Limited, the Dr. Jonathan Beare Family Office, and Goldby, Compton and Mackelvie Durban. Mr. Mun-Gavin earned a certificate in the theory of accountancy at University of Natal. We believe that Mr. Mun-Gavin is well-qualified to serve as a director due to his extensive finance experience and analytical background.
Term of Office
Each director serves until our next annual meeting or until his or her successor is duly elected and qualified. Each executive officer is elected by our board of directors and serves at its discretion.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our officers, directors, and persons who own more than ten percent of a registered class of our equity securities to file reports of ownership and changes in ownership with the SEC and to furnish the Company with copies of all Section 16(a) forms they file. Our review of copies of the Section 16(a) reports filed to report transactions occurring during the fiscal year ended December 31, 2025 indicates that all filing requirements applicable to our officers, directors, and greater than ten percent beneficial owners were complied with, except that late Form 3s were filed for each of Andrew Ritter, David Mun-Gavin and Hilary Kramer upon becoming insiders.
Audit Committee and Audit Committee Financial Expert
Our Audit Committee currently consists of Mr. Casamento (Chairman), Ms. Kramer and Mr. Mun-Gavin. Our Board of Directors has determined that all current and prospective members of our Audit Committee are independent under the Nasdaq listing rules and Rule 10A-3(b)(1) of the Exchange Act. Our Board of Directors has determined that Mr. Casamento is an audit committee financial expert, as defined in Item 407(d)(5) of Regulation S-K, and that each member of our Audit Committee is able to read and understand fundamental financial statements and has substantial business experience that results in such member’s financial sophistication. Accordingly, our Board of Directors believes that each member of our Audit Committee has sufficient knowledge and experience necessary to fulfill such member’s duties and obligations on our Audit Committee.
Code of Conduct and Ethics
We have adopted a written code of conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. A current copy of the code is posted on the corporate governance section of our website, which is located at www.gtbiopharma.com. If we make any substantive amendments to, or grant any waivers from, the code of conduct and ethics for any officer or director, we will disclose the nature of such amendment or waiver on our website or in a current report on Form 8-K.
Insider Trading Policy
We adopted an insider trading policy that governs the purchase, sale and/or disposition of our securities by our directors, officers, employees and consultants. We believe the insider trading policy is reasonably designed to promote compliance with insider trading laws, rules and regulations, and applicable Nasdaq listing standards. A copy of the insider trading policy is filed as Exhibit 19.1 to this Annual Report.
Clawback Policy
We have adopted a compensation recovery policy that is compliant with the Nasdaq Listing Rules, as required by the Dodd-Frank Act.
| 67 |
ITEM 11. EXECUTIVE COMPENSATION
Compensation of Executive Officers
Summary Compensation Table
The following table summarizes all compensation for the last two fiscal years awarded to, earned by, or paid to our Chief Executive Officer (principal executive officer) and our two most highly compensated executive officers other than our CEO during 2025.
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Stock Awards ($) | Option Awards (1) ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||
| Michael Breen | 2025 | 641,020 | 585,058 | – | 342,000 | (2) | 159,569 | (3) | 1,727,646 | |||||||||||||||||||
| Chairman, Chief Executive Officer, and President | 2024 | 556,200 | 417,150 | – | – | 213,622 | (4) | 1,186,972 | ||||||||||||||||||||
| Alan Urban | 2025 | 393,750 | 157,500 | – | 113,000 | (5) | 75,060 | (6) | 739,310 | |||||||||||||||||||
| Chief Financial Officer | 2024 | 218,750 | 87,500 | – | 40,681 | (7) | 30,851 | (8) | 377,782 | |||||||||||||||||||
| (1) | This column represents option awards computed in accordance with FASB ASC Topic 718, excluding the effect of estimated forfeitures related to service-based vesting conditions. For additional information on the valuation assumptions with respect to the option grants, refer to Note 1 of our financial statements in this Annual Report. These amounts do not correspond to the actual value that will be recognized by the named executives from these awards. |
| (2) | Represents the aggregate grant date fair value of options to purchase 300,000 shares of common stock issued on August 19, 2025, with 1/3rd of the shares full vested, 1/3rd of the shares vesting quarterly over 2025, and 1/3rd of the shares vesting quarterly over 2026, subject to acceleration. |
| (3) | Other compensation for 2025 includes $75,996 in paid time off payout, $60,000 in medical insurance, life insurance and long-term disability insurance premiums, and $23,573 in employer pension contributions. |
(4) |
Other compensation for 2024 includes $131,374 in paid time off payout, $60,000 in medical insurance, life insurance and long-term disability insurance premiums, and $22,248 in employer pension contributions. |
| (5) | Represents the aggregate grant date fair value of options to purchase 100,000 shares of common stock issued on August 19, 2025, with 1/2 of the shares vesting quarterly over 2025, and 1/2 of the shares vesting quarterly over 2026, subject to acceleration. |
| (6) | Other compensation for 2025 includes $61,060 in medical insurance premiums, and $14,000 employer 401(k) contributions. |
| (7) | Represents the aggregate grant date fair value of options to purchase 23,335 shares of common stock issued on October 17, 2024, with 1/36th of the shares vesting on the monthly anniversary of June 3, 2024 until fully vested, subject to acceleration. |
(8) |
Other compensation for 2024 includes $22,453 in medical insurance premiums, and $8,398 employer 401(k) contributions. |
Employment Agreements
The Company is party to employment agreements with both Michael Breen and Alan Urban, each of which are described below. The Company does not currently have employment agreements with any of its directors.
| 68 |
Michael Breen, Chairman, Chief Executive Officer and President
On December 31, 2021, the Company entered into an employment agreement with Mr. Breen, effective November 8, 2021, and amended on June 17, 2022, February 20, 2023, and August 26, 2025. The employment agreement shall automatically renew for successive two-year periods effective April 29, 2025, unless and until either party provides ninety (90) days’ advance written notice prior to applicable renewal term. Effective January 1, 2026, Mr. Breen will receive an annual base salary of $613,210, reimbursement for executive life insurance premiums, a pension contribution equal to 4% of Mr. Breen’s gross annual salary, and right to receive any benefit and participate in any benefit plan generally available to the most senior level of employees of the Company. Mr. Breen is eligible to participate in our performance bonus plan or as otherwise determined by our Compensation Committee, with a target annual bonus of 75% of his annual base salary with a minimum guaranteed performance bonus of 25% of base salary. Mr. Breen is also designated for election to our Board of Directors during the term of his employment agreement.
Upon the termination of Mr. Breen’s services for any reason, Mr. Breen will receive his accrued but unpaid salary and vacation pay through the date of termination and any other benefits accrued to him under any benefit plans outstanding at such time, and the reimbursement of documented, unreimbursed expenses incurred prior to such date. Upon our termination of Mr. Breen’s services without cause (as defined in the his services agreement) or upon Mr. Breen’s termination of his employment for good reason (as defined in his services agreement) prior to the end of the term of his services agreement, Mr. Breen shall also receive (i) a lump sum payment equal to the greater of the amount of his annual base salary (at the then-current rate) that he would have earned through the end of the term of the agreement, and 50% of his annual base salary, plus (ii) a lump sum payment equal to the greater of the bonus paid or payable to Mr. Breen for the immediately preceding year, and the target bonus under our performance bonus plan, if any, in effect during the immediately preceding year, plus (iii) monthly reimbursement for the cost of medical, life and disability insurance coverage at a level equivalent to that provided by our company for a period of the earlier of (a) one year and (b) the time Mr. Breen begins alternative services wherein said insurance coverage is available and offered to Mr. Breen.
Alan Urban, Chief Financial Officer
On June 7, 2024, the Company entered into an Employment Agreement with Mr. Urban (the “Employment Agreement”), effective from June 3, 2024 (the “Effective Date”). The Employment Agreement shall automatically renew for successive one-year periods unless and until either party provides sixty (60) days’ advance written notice prior to applicable renewal term. Effective January 1, 2025, Mr. Urban received an annual base salary of $393,750 and is eligible to earn an annual discretionary bonus of up to 40% of his annual base salary each calendar year during the term, subject to the achievement of applicable Company and individual performance goals, as determined in the Company’s sole discretion. Mr. Urban is eligible to receive a stock award of the common stock following the three months after the Effective Date. The Employment Agreement further provides that Mr. Urban will be eligible to receive any benefit and participate in any benefit plan generally available to employees of the Company.
The Company may terminate the Employment Agreement without Cause (as such term is defined in the Employment Agreement) at any time and Mr. Urban may terminate his employment for Good Reason (as such term is defined in the Employment Agreement) at any time, in each case, subject to one months’ notice by the terminating party, or pay in lieu thereof, if terminated by the Company without Cause. Upon a termination of Mr. Urban’s employment by the Company without Cause or by Mr. Urban for Good Reason, Mr. Urban will be entitled to receive (i) for a termination or resignation that occurs during the first six months following the Effective Date, a cash severance equal to two (2) months of Mr. Urban’s then current Annual Base Salary (as such term is defined in the Employment Agreement) or (ii) for a termination or resignation that occurs any time thereafter, a cash severance equal to five (5) months of Mr. Urban’s then current Annual Base Salary, in either case less deductions and withholding required by law, payable in a lump sum within seventy (70) days of the termination of employment (or such shorter period as may allow the severance payment to be exempt from Code Section 409A). Upon a termination of Mr. Urban’s employment by the Company for Cause or by Mr. Urban without Good Reason, Mr. Urban will be entitled to the Accrued Amounts (as such term is defined in the Employment Agreement). The Employment Agreement also contains certain non-disclosure covenants that apply during his employment and thereafter.
| 69 |
Outstanding Equity Awards at Fiscal Year End
The following table sets forth information regarding outstanding stock options for each named executive officer as of December 31, 2025.
| Option Awards | ||||||||||||||
| Name | Number of securities underlying unexercised options exercisable (#) | Number of securities underlying unexercised options unexercisable (#) | Option exercise price ($) | Option expiration date | ||||||||||
| Michael Breen | 1,666 | (1) | — | 74.40 | 7/14/2032 | |||||||||
| 16,666 | (1) | — | 25.50 | 1/27/2033 | ||||||||||
| 200,000 | (2) | 100,000 | (2) | 1.33 | 8/19/2035 | |||||||||
| Alan Urban | 11,668 | (3) | 11,667 | (3) | 2.11 | 10/17/2034 | ||||||||
| 50,000 | (4) | 50,000 | (4) | 1.33 | 8/19/2035 | |||||||||
| (1) | Consists of options granted to Mr. Breen as compensation for service on our Board of Directors. |
|
(2) |
Consists of options granted to Mr. Breen that vest as follows: 1/3rd of the shares full vested, 1/3rd of the shares vesting quarterly over 2025, and 1/3rd of the shares vesting quarterly over 2026, subject to acceleration. |
| (3) | Consists of options granted to Mr. Urban that vest as follows: 1/2 of the shares vesting quarterly over 2025, and 1/2 of the shares vesting quarterly over 2026, subject to acceleration. |
| (4) | Consists of options granted to Mr. Urban that vest as follows: 1/36th of the shares vest on the monthly anniversary of June 3, 2024 until fully vested, subject to acceleration. |
Compensation of Directors
The following table presents information regarding compensation awarded or paid to our non-employee directors for our fiscal year ended December 31, 2025, for the services rendered by them to the Company in all capacities.
| Name | Fees earned or paid in cash(7) ($) | Stock awards ($) | Option Awards (1) ($) | Total ($) | ||||||||||||
| Charles J. Casamento | 65,000 | — | 66,880 | (2) | 131,880 | |||||||||||
| Hilary Kramer | 42,250 | — | 33,440 | (3) | 75,690 | |||||||||||
| David C. Mun-Gavin | 36,111 | — | 33,440 | (3) | 69,551 | |||||||||||
| Andrew Ritter(4) | 5,778 | — | — | 5,778 | ||||||||||||
| Rajesh Shrotriya, M.D. (5) | 23,834 | — | — | 23,834 | ||||||||||||
| Bruce Wendel (6) | 22,931 | — | — | 22,931 | ||||||||||||
| (1) | This column represents option awards computed in accordance with FASB ASC Topic 718, excluding the effect of estimated forfeitures related to service-based vesting conditions. For additional information on the valuation assumptions with respect to the option grants, refer to Note 1 of our financial statements in this Annual Report. These amounts do not correspond to the actual value that will be recognized by the non-employee directors from these awards. |
| (2) | Represents the aggregate grant date fair value of fully vested options to purchase 62,500 shares of common stock at $1.33 per share issued on August 19, 2025. |
| (3) | Represents the aggregate grant date fair value of fully vested options to purchase 31,250 shares of common stock at $1.33 per share issued on August 19, 2025. |
| (4) | Andrew Ritter served as a director from May 8, 2025 to June 9, 2025, when he resigned as a member of the Board. |
| (5) | Rajesh Shrotriya, M.D. resigned as a member of the Board on May 12, 2025. |
| (6) | Bruce Wendel resigned as a member of the Board on May 7, 2025. |
| (7) | Prorated based on start and end date. |
During fiscal 2025, our non-employee directors received annual cash compensation in the amount of $50,000 for service on our Board of Directors, and annual compensation of $5,000 per committee for service on our Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee.
Indemnification of Directors and Executive Officers and Limitation of Liability
We have entered into indemnification agreements with each of our current directors and certain key employees. The indemnification agreements, our Charter and Bylaws require us to indemnify our current and former directors and officers to the fullest extent permitted by Delaware law.
Policies and Practices Related to the Grant of Certain Equity Awards Close in Time to the Release of Material Nonpublic Information
While
we do not have a formal written policy in place with regard to the timing of awards of options in relation to the disclosure of material
nonpublic information,
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information, as of February 23, 2026, with respect to the holdings of (1) each person who is the beneficial owner of more than five percent of our common stock, (2) each of our directors and director nominees, (3) each named executive officer, and (4) all of our directors and executive officers as a group.
| 70 |
Beneficial ownership of our common stock is determined in accordance with the rules of the Securities and Exchange Commission and includes any shares of common stock over which a person exercises sole or shared voting or investment powers, or of which a person has a right to acquire ownership at any time within 60 days of February 23, 2026. Except as otherwise indicated, and subject to applicable community property laws, the persons named in this table have sole voting and investment power with respect to all shares of common stock held by them. The address of each director and officer is 505 Montgomery Street, 10th Floor, San Francisco, California 94111. Applicable percentage ownership in the following table is based on 31,553,892 shares of common stock outstanding as of February 23, 2026 plus, for each person, any securities that person has the right to acquire within 60 days of February 23, 2026.
| Name of Beneficial Owner | Number of Shares Beneficially Owned | Percentage of Shares Outstanding | ||||||
| Five Percent or Greater Stockholders: | ||||||||
| Bristol Investment Fund, Ltd (1) (2) | 3,146,380 | 9.99 | % | |||||
| Five Narrow Lane, L.P. (1) (3) | 3,146,380 | 9.99 | % | |||||
| The Hewlett Fund LP (4) | 2,014,542 | 6.40 | % | |||||
| Intracoastal Capital LLC (5) | 3,146,380 | 9.99 | % | |||||
| Robert A. Marzilli (6) | 3,146,380 | 9.99 | % | |||||
| Executive Officers and Directors: | ||||||||
| Michael Breen (7) | 245,205 | — | % | |||||
| Alan Urban (8) | 61,668 | — | % | |||||
| Charles J. Casamento (9) | 79,166 | — | % | |||||
| Hilary Kramer. (10) | 31,250 | — | % | |||||
| David C. Mun-Gavin (10) | 31,250 | — | % | |||||
| Directors and officers as a group (5 persons) (11) | 131,875 | — | % | |||||
| (1) | In accordance with Rule 13d-3(d) under the Exchange Act, we have excluded from the number of shares of common stock beneficially owned all of the shares that a Facility Investor may be required to purchase under the Common Shares Purchase Agreement. The issuance of such shares is solely at our discretion and is subject to conditions contained in the Common Shares Purchase Agreement, the satisfaction of which are entirely outside of the Facility Investors’ control. Furthermore, the VWAP Purchases (as defined in the Common Shares Purchase Agreement) of our common stock under the Common Shares Purchase Agreement are subject to certain agreed upon maximum amount limitations set forth in the Common Shares Purchase Agreement, including the contractually stipulated 4.99% or 9.99% blocker, as applicable. |
| (2) | The address for Bristol Investment Fund, Ltd. (“BIF”) is Citco Trustees (Cayman) Limited, 89 Nexus Way, Camana Bay, PO Box 311063, Grand Cayman KY1-1205, Cayman Islands. Paul Kessler, as manager of Bristol Capital Advisors, LLC, the investment advisor to BIF, has voting and investment control over the securities held by BIF. Mr. Kessler, as manager of Bristol and Hailstone, has voting and investment control over the securities held by Bristol and Hailstone. Mr. Kessler disclaims beneficial ownership of these securities except to the extent of his pecuniary interest therein. Bristol reported to the Company that, as of January 12, 2026, shares beneficially owned consists of (i) 884,520 shares of common stock by BIF, and (ii) 2,261,860 shares of common stock issuable upon the conversion of Series L Preferred Stock and/or exercise of the Common Stock Warrants or Vesting Warrants held by BIF within 60 days of February 23, 2026. The shares beneficially owned reflects the application of a contractually stipulated 9.99% blocker provision. |
| (3) | The address for 5NL is 510 Madison Avenue, Suite 1400, New York, New York 10022. Each of Arie Rabinowitz and Joseph Hammer may be deemed to have investment discretion and voting power over the shares held by FNL. Each of Messrs. Rabinowitz and Hammer disclaims any beneficial ownership of these shares except to the extent of his pecuniary interest therein. 5NL reported to the Company that, as of January 12, 2026, shares beneficially owned consists of (i) 131,817 shares of common stock, and (ii) 3,014,563 shares of common stock issuable upon the conversion of Series L Preferred Stock and/or exercise of the Common Stock Warrants or Vesting Warrants held by 5NL within 60 days of February 23, 2026. The shares beneficially owned reflects the application of a contractually stipulated 9.99% blocker provision. |
| (4) | The address for The Hewlett Fund LP is 100 Merrick Road, Suite 400W, Rockville Centre, New York 11570. Martin Chopp has voting and investment control over the securities held by The Hewlett Fund LP. The Hewlett Fund LP reported to the Company that, as of January 12, 2026, shares beneficially owned consists of (i) 14,974 shares of common stock, and (ii) 1,999,568 shares of common stock issuable upon the conversion of Series L Preferred Stock and/or exercise of the Common Stock Warrants or Vesting Warrants held by The Hewlett Fund LP within 60 days of February 23, 2026. The shares beneficially owned reflects the application of a contractually stipulated 9.99% blocker provision. |
| 71 |
| (5) | The address for Intracoastal Capital LLC (“Intracoastal”) is 245 Palm Trail, Delray Beach, Florida 33843. Mitchell P. Kopin (“Mr. Kopin”) and Daniel B. Asher (“Mr. Asher”), each of whom are managers of Intracoastal, have shared voting control and investment discretion over the securities reported herein that are held by Intracoastal. As a result, each of Mr. Kopin and Mr. Asher may be deemed to have beneficial ownership (as determined under Section 13(d) of the Exchange Act) of the securities reported herein that are held by Intracoastal. Intracoastal reported to the Company that, as of January 12, 2026, shares beneficially owned consists of (i) 18,319 shares of common stock, and (ii) 3,128,061 shares of common stock issuable upon the conversion of Series L Preferred Stock and/or exercise of the Common Stock Warrants or Vesting Warrants held by Intracoastal within 60 days of February 23, 2026. The shares beneficially owned reflects the application of a contractually stipulated 9.99% blocker provision. |
| (6) | The address for Robert A. Marzilli is 457 Sunset Beach Rd., Richmond Hill, Ontario, L4E 3J3, Canada. This information is based on information known to the company through a non-objecting beneficial ownership report (the “NOBO Report”) as of January 31, 2026. Mr. Marzilli has not provided or verified the information appearing on the NOBO Report, and so this information may not be accurate for a number of reasons, including, but not limited to, if Mr. Marzilli has divested such ownership through private contractual or other means not reflected in the NOBO Report, or is the beneficial owner of other shares not disclosed in the NOBO Report. Shares beneficially owned consists of (i) 700,000 shares of common stock, and (ii) 2,446,380 shares of common stock issuable upon the conversion of Series L Preferred Stock and/or exercise of the Common Stock Warrants or Vesting Warrants held by Mr. Marzilli within 60 days of February 23, 2026. The shares beneficially owned reflects the application of a contractually stipulated 9.99% blocker provision. |
| (7) | Consists of 26,873 shares of common stock, and includes shares underlying options to purchase 1,666 shares of common stock at $74.40 per share, 16,666 shares of common stock at $25.50 per share, and 200,000 shares of common stock at $1.33 per share. |
| (8) | Includes shares underlying options to purchase 11,668 shares of common stock at $2.11 per share, and 50,000 shares of common stock at $1.33 per share. |
(9) |
Includes shares underlying options to purchase 16,666 shares of common stock at $10.50 per share, and 62,500 shares of common stock at $1.33 per share. |
| (10) | Includes shares underlying options to purchase 31,250 shares of common stock at $1.33 per share. |
| (11) | Includes shares underlying options to purchase 421,666 shares of common stock. |
Equity Compensation Plan Information
In April 2014 we established the 2014 Stock Incentive Plan (the “2014 Plan”) and in April 2022 we established the 2022 Omnibus Incentive Plan (as amended, the “2022 Plan”), collectively (the “Plans”). The Plans were approved by our Board of Directors and stockholders. The purpose of the Plans is to grant stock and options to purchase our common stock, and other incentive awards, to our employees, directors and key consultants. On July 24, 2025, stockholders voted to increase the maximum number of shares of common stock that may be issued pursuant to awards granted under the 2022 Plan by 583,334 shares. Pursuant to the increase, the maximum number of shares of common stock that may be issued pursuant to awards granted under the 2022 Plan is 750,000. Following the adoption of the 2022 Plan by our stockholders, we only grant incentive awards under the 2022 Plan. The shares of our common stock underlying cancelled and forfeited awards issued under the 2022 Plan may again become available for grant under the 2022 Plan. As of December 31, 2025, there were 597,550 stock options outstanding and 25,935 shares of restricted stock granted in prior years under the 2022 Plan, which left 126,515 shares available for grant under the 2022 Plan. All outstanding incentive stock award grants prior to the adoption of the 2022 Plan were made under the 2014 Plan, and all incentive stock award grants after the adoption of the 2022 Plan have been and will be made under the 2022 Plan. The following table provides information as of December 31, 2025 with respect to the Plans:
| Plan category | Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | |||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by stockholders (2014 Plan) | — | — | ||||||||||
| Equity compensation plans approved by stockholders (2022 Plan) | 597,550 | $ | 4.23 | 126,516 | ||||||||
| Total | 597,550 | 126,516 | ||||||||||
| 72 |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Transactions with Officers, Directors and greater than 5% stockholders
Other than listed below, since January 1, 2024, there has not been, nor is there currently proposed, any transaction or series of similar transactions to which we were or will be a party:
| ● | in which the amount involved exceeds the lesser of $120,000 or one percent of the average of our total assets at year end for the last two completed fiscal years; and |
| ● | in which any director, executive officer, stockholder who beneficially owns more than 5% of our common stock or any member of their immediate family had or will have a direct or indirect material interest. |
PDPC Advisors Inc.
On June 30, 2025, the Company entered into an Advisory Agreement (the “Advisory Agreement”) with PDPC Advisors Inc. (“PDPC”), to perform certain advisory services. Under the Advisory Agreement cash payments amounting to $100,000 are to be paid in six equal installments beginning on July 1, 2025 and ending on December 31, 2025. In addition, upon execution of the Advisory Agreement, the Company issued to PDPC a pre-funded warrant to purchase 150,000 shares of common stock of the Company, which had a fair value of $537,000 at the time of issuance. The Advisory Agreement began on July 1, 2025 and terminates on June 30, 2026. PDPC is considered a related party as its CEO is an individual who has voting and investment control over an entity whose beneficial ownership exceeded 5% of the issued and outstanding shares of the Company’s common stock.
Cytovance Biologics, Inc.
In October 2020, the Company entered into a Master Services Agreement with Cytovance Biologics, Inc. (“Cytovance”), to perform biologic development and manufacturing services, and to produce and test compounds used in the Company’s potential product candidates. The Company subsequently executed numerous Statements of Work (“SOWs”) for the research and development of products for use in clinical trials.
On August 24, 2022, the Company entered into a Settlement and Investment Agreement with Cytovance that amended existing SOWs and allowed for future invoices to be settled in in a combination of cash and issuance of the Company’s common stock. The Agreement also set Cytovance’s beneficial ownership limitation at 4.9% of the issued and outstanding shares of the Company’s common stock.
On April 25, 2024, the Company entered into an Amendment to the Settlement and Investment Agreement with Cytovance that increased Cytovance’s beneficial ownership limitation to 9.9% of the issued and outstanding shares of the Company’s common stock. Cytovance was a related party as of the year ended December 31, 2024, but not as of the year ended December 31, 2025.
During the years ended December 31, 2025 and 2024, the Company recognized research and development expenses of $743,000 and $2,335,000, respectively and made cash payments amounting to $1,054,000 and $3,857,000, respectively to Cytovance. In addition, during the year ended December 31, 2025, the Company issued pre-funded warrants to purchase up to 326,251 shares of common stock exercisable at $0.0001 per share to settle accounts payable valued at approximately $847,000; and during the year ended December 31, 2024 the Company issued 127,597 shares of common stock to Cytovance to settle accounts payable valued at approximately $810,000.
As of December 31, 2025 the Company’s commitments in relation to unbilled and unaccrued SOWs and any related Change Orders from Cytovance for services that have not yet been rendered as of December 31, 2025, amounted to approximately $169,000.
Director Independence
We utilize the Nasdaq listing rules in determining whether a director is independent. The Nasdaq rules generally define an “independent director” as a person, other than an executive officer of a company or any other individual having a relationship which, in the opinion of the issuer’s board of directors, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Mr. Breen is not considered to be independent due to his role as an executive officer of the Company. The Board has determined that each of Mr. Casamento and Mr. Mun-Gavin and Ms. Kramer qualifies as an independent director, and that the Board currently consists of a majority of independent directors, as such term is defined under the Nasdaq rules. In making this determination, our Board considered the current and prior relationships, as applicable, that each of Mr. Casamento and Mr. Mun-Gavin and Ms. Kramer has with our Company and all other facts and circumstances our Board deemed relevant in determining their independence, including their beneficial ownership of our common stock.
| 73 |
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Summary of Principal Accounting Fees for Professional Services Rendered
Our independent registered public accounting firm is Weinberg & Company, P.A. 1925 Century Park E., Suite 1120, Los Angeles, CA 90067. PCAOB Auditor ID: 572. The following table presents the aggregate fees for professional audit services and other services rendered in the fiscal years ended December 31, 2025 and 2024.
| Year Ended December 31, 2025 | Year Ended December 31, 2024 | |||||||
| Audit Fees | $ | 214,198 | $ | 199,925 | ||||
| Audit-Related Fees | — | — | ||||||
| Tax Fees | 19,506 | 28,489 | ||||||
| All Other Fees | — | — | ||||||
| Total | $ | 233,704 | $ | 228,414 | ||||
Audit Fees consist of amounts billed for professional services rendered for the audit of our annual financial statements included in our Annual Reports on Form 10-K, and reviews of our interim financial statements included in our Quarterly Reports on Form 10-Q.
Audit-Related Fees consist of fees billed for professional services that are reasonably related to the performance of the audit or review of our financial statements but are not reported under “Audit Fees.”
Tax Fees consist of fees for professional services for tax compliance activities, including the preparation of federal and state tax returns and related compliance matters.
All Other Fees consists of amounts billed for services other than those noted above, including comfort letters, and consents.
The audit committee of our board of directors has considered whether the provision of the services described above for the fiscal years ended December 31, 2025 and 2024, is compatible with maintaining the auditor’s independence.
All audit and non-audit services that may be provided by our principal accountant to us shall require pre-approval by the audit committee of our Board. Further, our auditor shall not provide those services to us specifically prohibited by the SEC, including bookkeeping or other services related to the accounting records or financial statements of the audit client; financial information systems design and implementation; appraisal or valuation services, fairness opinion, or contribution-in-kind reports; actuarial services; internal audit outsourcing services; management functions; human resources; broker-dealer, investment adviser, or investment banking services; legal services and expert services unrelated to the audit; and any other service that the Public Company Accounting Oversight Board determines, by regulation, is impermissible.
| 74 |
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a)(1) Financial Statements.
The financial statements of GT Biopharma, Inc. and the independent registered public accounting firm’s report dated March 2, 2026, are incorporated by reference to Item 8 of this report.
(a)(2) and (c) Financial Statement Schedules
Not required.
(a)(3) and (b) Exhibits
EXHIBIT INDEX
| 75 |
| 76 |
| 77 |
++ Indicates management contract or compensatory plan.
* This certification shall not be deemed “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that Section, nor shall it be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act.
**The Registrant has omitted portions of this exhibit that are both not material and the type of information that the Registrant treats as private or confidential.
ITEM 16. FORM 10-K SUMMARY
Not applicable.
| 78 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
GT Biopharma, Inc.
| ||
| Dated: March 2, 2026 | By: | /s/ Michael Breen |
Michael Breen, Executive Chairman of the Board and Chief Executive Officer (Principal Executive Officer) | ||
GT Biopharma, Inc.
| ||
| Dated: March 2, 2026 | By: | /s/ Alan Urban |
| Alan Urban, Chief Financial Officer | ||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Michael Breen and Alan Urban, and each of them, as his true and lawful attorneys-in-fact and agents, with full power of substitution for him, and in his name in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, and any of them or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | Position | Date | ||
| /s/ Michael Breen | Executive Chairman of the Board and | March 2, 2026 | ||
| Michael Breen | Chief Executive Officer (Principal Executive Officer) | |||
|
||||
| /s/ Alan Urban | Chief Financial Officer | March 2, 2026 | ||
| Alan Urban | (Principal Financial and Accounting Officer) | |||
|
||||
| /s/ Charles Casamento | Director | March 2, 2026 | ||
| Charles Casamento | ||||
|
||||
| /s/ Hilary Kramer | Director | March 2, 2026 | ||
| Hilary Kramer | ||||
|
||||
| /s/ David Mun-Gavin | Director | March 2, 2026 | ||
| David Mun-Gavin |
| 79 |